
Validation of a Novel Cuproptosis–Related Prognostic Gene Marker and Differential Expression Associated with Lung Adenocarcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition of LUAD Gene Expression Profile
2.2. Differential Expression of CRGs in LUAD
2.3. Gene Network and Enrichment Analysis of CRGs
2.4. Prognostic Analysis and Clinical Staging of CRGs
2.5. Construction of the Prognostic Signature of CRGs in LUAD
2.6. RNA Modified Genes and miRNA Analysis
2.7. Analysis of Correlation with Immune Infiltration
2.8. Single–Cell–Type Analysis and Immunohistochemical Staining
3. Results
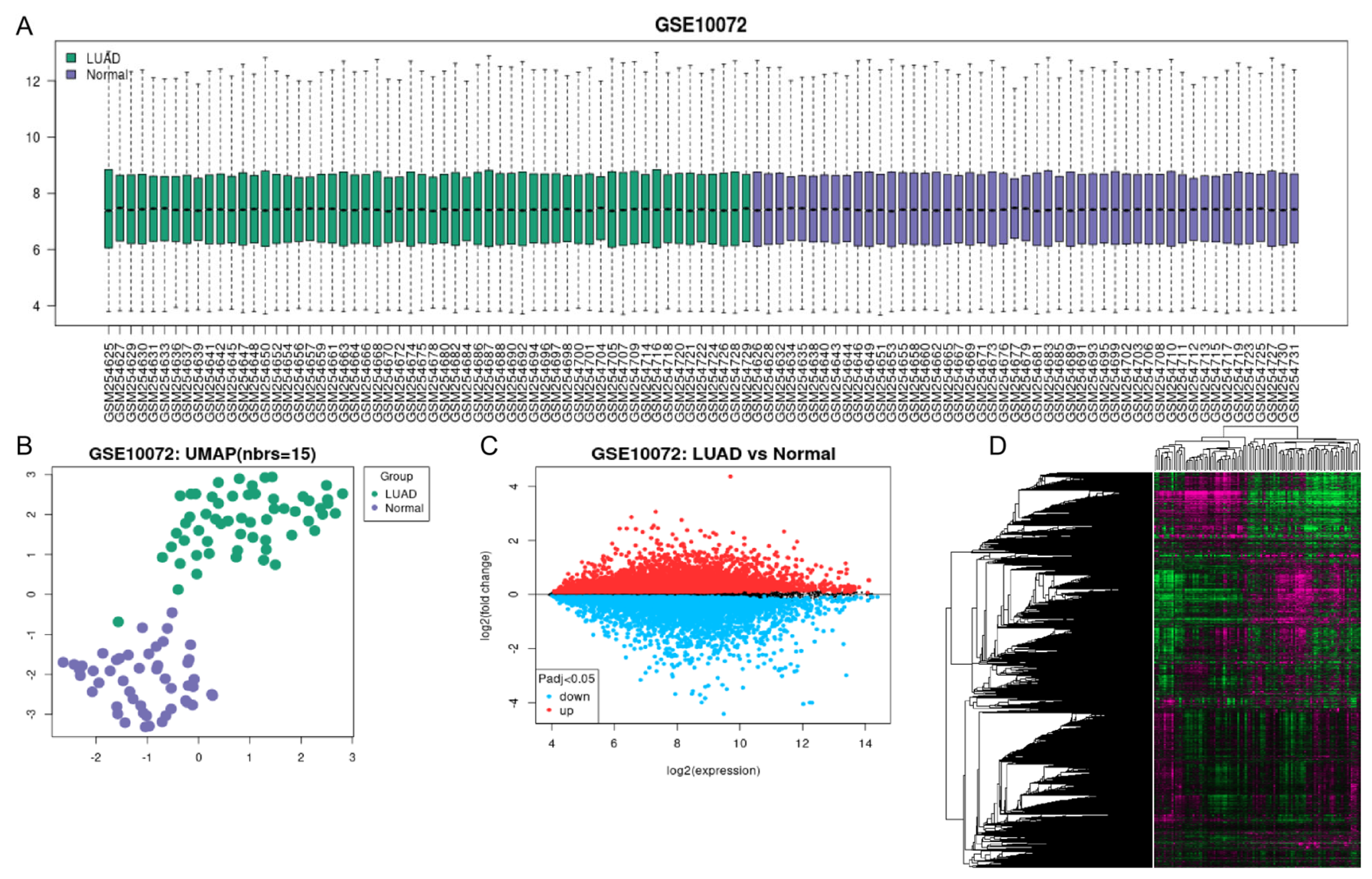
3.1. Data Acquisition of LUAD Genes Expression Profile
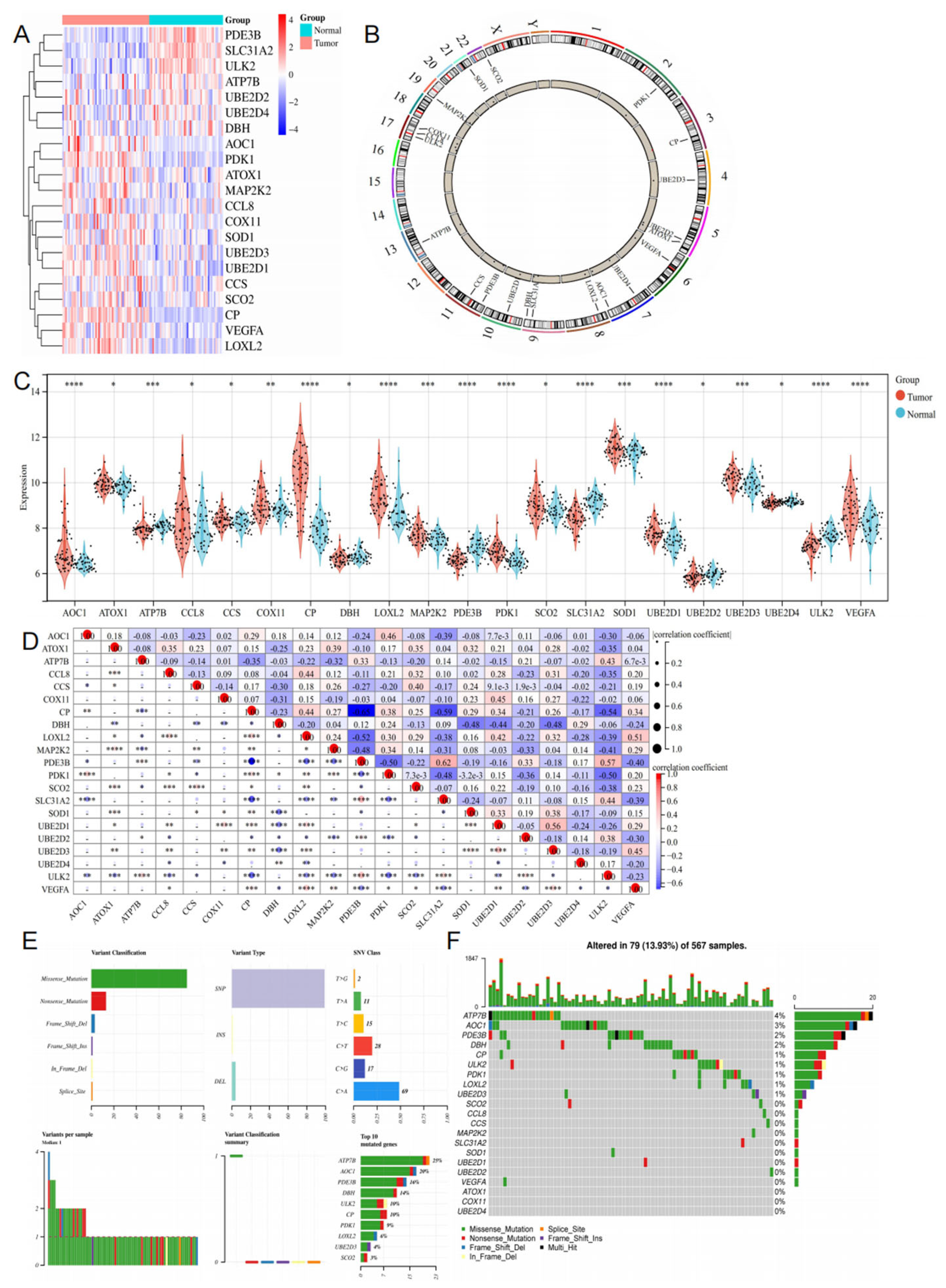
3.2. Differential Expression of CRGs in LUAD
3.3. Functional Enrichment and Protein–Protein Interaction Analysis of CRGs
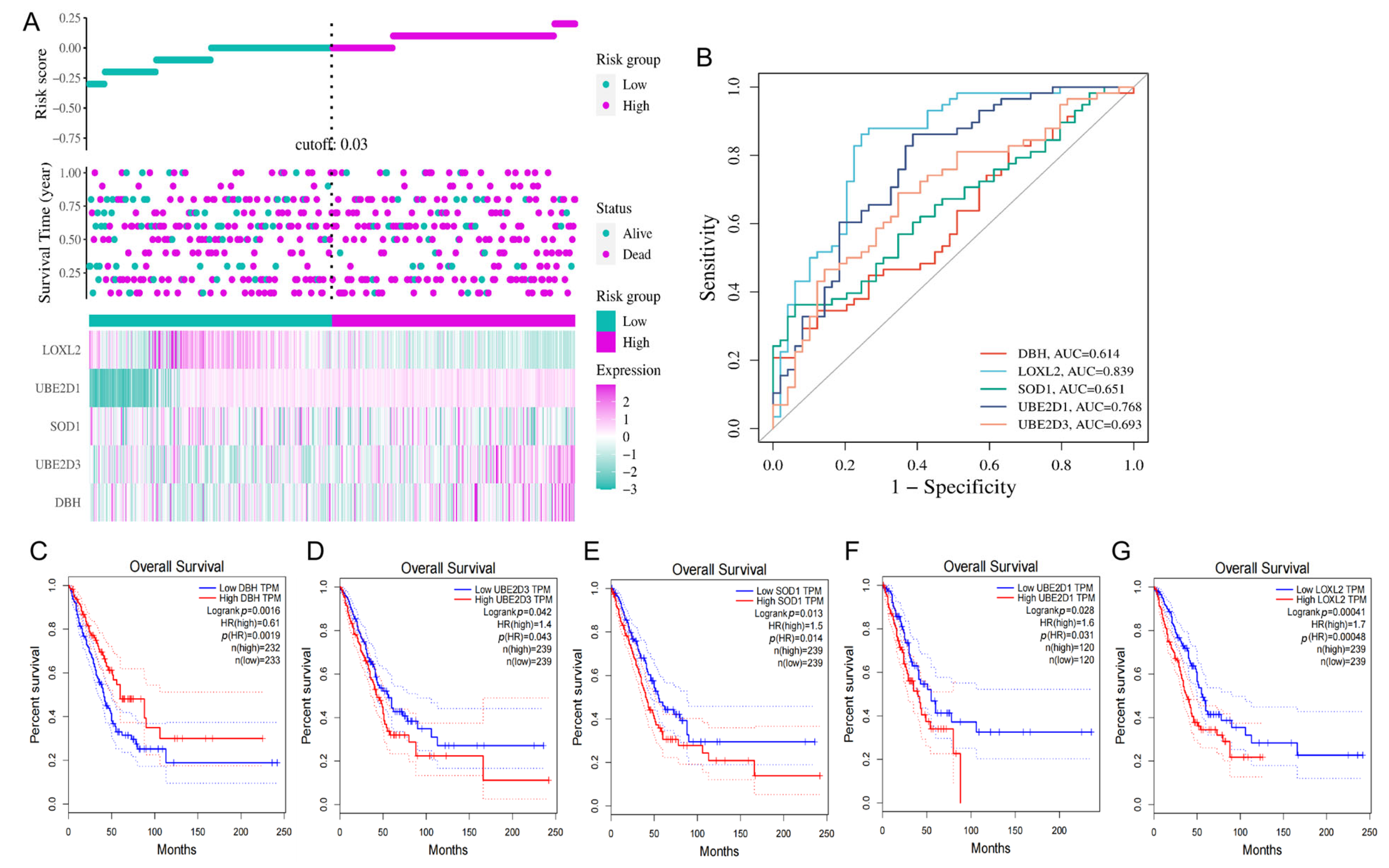
Prognostic Analysis and Clinical Staging of CRGs
3.4. Construction of the Prognostic Signature of CRGs in LUAD
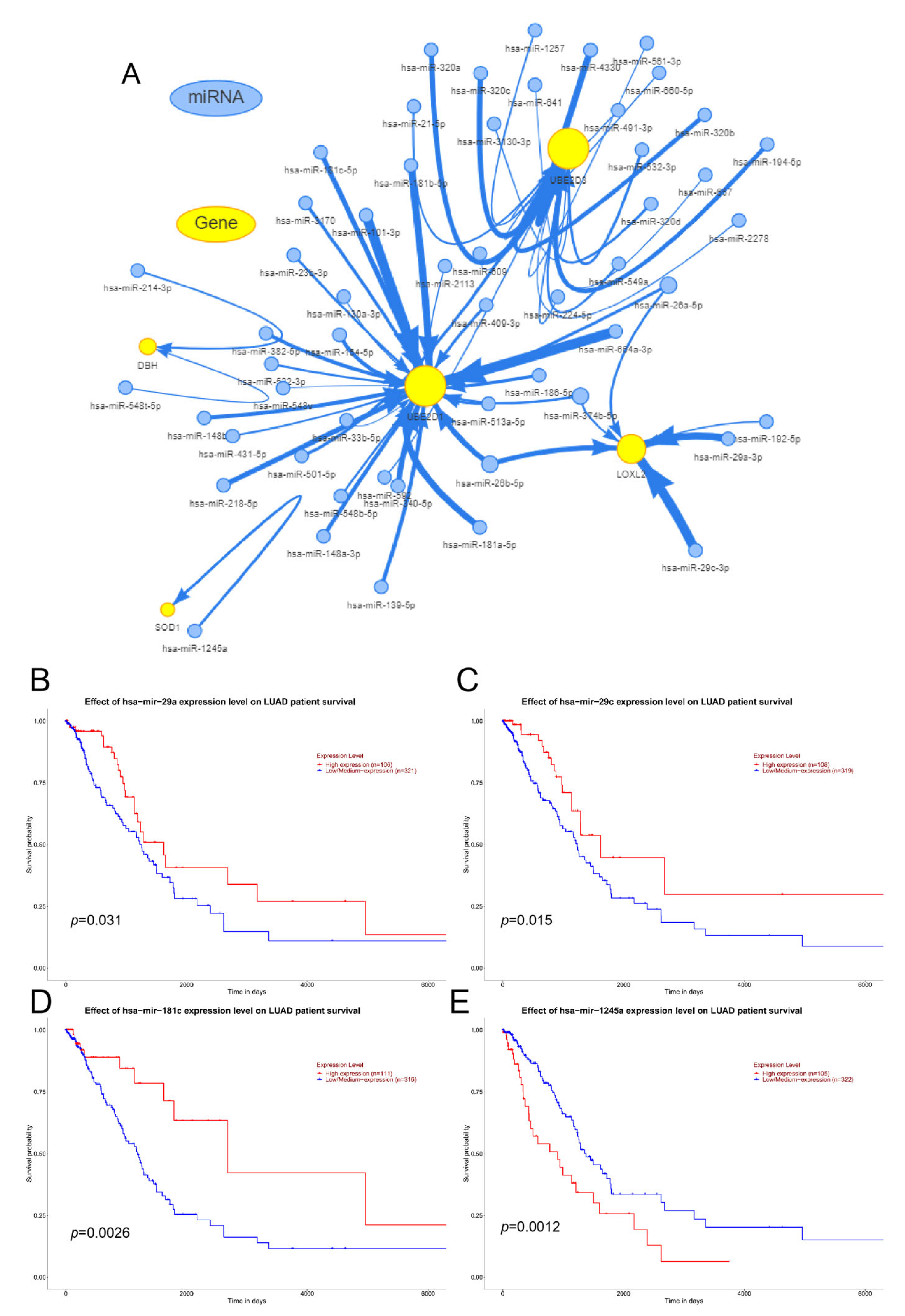
3.5. RNA Modified Genes and miRNA Analysis
3.6. Correlation between the Expression of CRGs and Levels of Immune Infiltration in LUAD
3.7. Single–Cell–Type Analysis and Immunohistochemical Staining
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruiz-Cordero, R.; Devine, W.P. Targeted Therapy and Checkpoint Immunotherapy in Lung Cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Kadara, H.; Choi, M.; Zhang, J.; Parra, E.; Rodriguez-Canales, J.; Gaffney, S.; Zhao, Z.; Behrens, C.; Fujimoto, J.; Chow, C.; et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann. Oncol. 2018, 29, 1072. [Google Scholar] [CrossRef]
- Postmus, P.E.; Kerr, K.M.; Oudkerk, M.; Senan, S.; Waller, D.A.; Vansteenkiste, J.; Escriu, C.; Peters, S.; ESMO Guidelines Committee. Early and Locally Advanced Non-Small-Cell Lung Cancer (NSCLC): ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up. Ann. Oncol. 2017, 28, iv1–iv21. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, J.-B.; Hou, L.-K.; Yu, F.; Zhang, J.; Wu, W.; Tang, X.-M.; Sun, F.; Lu, H.-M.; Deng, J.; et al. Liquid biopsy in lung cancer: Significance in diagnostics, prediction, and treatment monitoring. Mol. Cancer 2002, 21, 25. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.C.; Hidalgo, L.H.; Moreno, E.; Correa, C.P.; de Vega, V.M. Papel de las técnicas de imagen en la nueva clasificación TNM del carcinoma broncogénico no microcítico [Role of imaging techniques in the TNM classification of non-small cell broncho-genic carcinoma]. Radiologia 2012, 54, 306–320. [Google Scholar] [CrossRef]
- Gajewski, T.F. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non–T-Cell–Inflamed Tumor Microenvironment. Semin. Oncol. 2015, 42, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer 2022, 22, 102–113. [Google Scholar] [CrossRef]
- Wang, L.; Bammler, T.K.; Beyer, R.P.; Gallagher, E.P. Copper-Induced Deregulation of microRNA Expression in the Zebrafish Olfactory System. Environ. Sci. Technol. 2013, 47, 7466–7474. [Google Scholar] [CrossRef]
- Blockhuys, S.; Wittung-Stafshede, P. Roles of Copper-Binding Proteins in Breast Cancer. Int. J. Mol. Sci. 2017, 18, 871. [Google Scholar] [CrossRef] [PubMed]
- Kardos, J.; Héja, L.; Simons, Á.; Jablonkai, I.; Kovács, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal. 2018, 16, 1–22. [Google Scholar] [CrossRef]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef]
- Shanbhag, V.; Jasmer-McDonald, K.; Zhu, S.; Martin, A.L.; Gudekar, N.; Khan, A.; Ladomersky, E.; Singh, K.; Weisman, G.A.; Petris, M.J. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc. Natl. Acad. Sci. USA 2019, 116, 6836–6841. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; He, Y.; Yu, Z.; Hu, J.; Xiao, Z.; Zu, X.; Li, Z.; Li, H. Cuproptosis-related modification patterns depict the tumor microenvironment, precision immunotherapy, and prognosis of kidney renal clear cell carcinoma. Front. Immunol. 2022, 13, 933241. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Heagerty, P.J.; Zheng, Y. Survival Model Predictive Accuracy and ROC Curves. Biometrics 2005, 61, 92–105. [Google Scholar] [CrossRef]
- Liu, C.-J.; Hu, F.-F.; Xia, M.-X.; Han, L.; Zhang, Q.; Guo, A.-Y. GSCALite: A web server for gene set cancer analysis. Bioinformatics 2018, 34, 3771–3772. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Sjöstedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef]
- Teng, P.-C.; Liang, Y.; Yarmishyn, A.A.; Hsiao, Y.-J.; Lin, T.-Y.; Lin, T.-W.; Teng, Y.-C.; Yang, Y.-P.; Wang, M.-L.; Chien, C.-S.; et al. RNA Modifications and Epigenetics in Modulation of Lung Cancer and Pulmonary Diseases. Int. J. Mol. Sci. 2021, 22, 10592. [Google Scholar] [CrossRef]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef]
- Theler, D.; Dominguez, C.; Blatter, M.; Boudet, J.; Allain, F.H.-T. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: A reader of methylated RNA. Nucleic Acids Res. 2014, 42, 13911–13919. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cheng, W.; Ren, X.; Wang, Z.; Liu, X.; Li, G.; Han, S.; Jiang, T.; Wu, A. Tumor Purity as an Underlying Key Factor in Glioma. Clin. Cancer Res. 2017, 23, 6279–6291. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Z.; Chen, R.; Fang, C.; Zhang, C.; Ji, M.; Yang, X. Immunotherapy-based combination strategies for treatment of EGFR-TKI-resistant non-small-cell lung cancer. Futur. Oncol. 2022, 18, 1757–1775. [Google Scholar] [CrossRef] [PubMed]
- Michalczyk, K.; Cymbaluk-Płoska, A. The Role of Zinc and Copper in Gynecological Malignancies. Nutrients 2020, 12, 3732. [Google Scholar] [CrossRef]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in Neurobiology: Probing Their Chemistry and Biology with Molecular Imaging. Chem. Rev. 2008, 108, 1517–1549. [Google Scholar] [CrossRef]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta BBA Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Liao, Y.; Zhao, J.; Bulek, K.; Tang, F.; Chen, X.; Cai, G.; Jia, S.; Fox, P.L.; Huang, E.; Pizarro, T.T.; et al. Inflammation mobilizes copper metabolism to promote colon tumorigenesis via an IL-17-STEAP4-XIAP axis. Nat. Commun. 2020, 11, 900. [Google Scholar] [CrossRef]
- Paredes, S.E.Y.; Almeida, L.Y.; Trevisan, G.L.; Polanco, X.B.J.; Silveira, H.A.; Silva, E.V.; Segato, R.A.B.; da Silva, L.A.B.; Chahud, F.; León, J.E. Immunohistochemical characterization of immune cell infiltration in paediatric and adult Langerhans cell histiocytosis. Scand. J. Immunol. 2020, 92, e12950. [Google Scholar] [CrossRef]
- Chen, F.; Han, B.; Meng, Y.; Han, Y.; Liu, B.; Zhang, B.; Chang, Y.; Cao, P.; Fan, Y.; Tan, K. Ceruloplasmin correlates with immune infiltration and serves as a prognostic biomarker in breast cancer. Aging 2021, 13, 20438–20467. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.M.F.; Hsin, M.K. New T1 classification. Gen. Thorac. Cardiovasc. Surg. 2020, 68, 665–671. [Google Scholar] [CrossRef]
- Travis, W.D.; Asamura, H.; Bankier, A.A.; Beasley, M.B.; Detterbeck, F.; Flieder, D.B.; Goo, J.M.; MacMahon, H.; Naidich, D.; Nicholson, A.G.; et al. The IASLC Lung Cancer Staging Project: Proposals for Coding T Categories for Subsolid Nodules and Assessment of Tumor Size in Part-Solid Tumors in the Forthcoming Eighth Edition of the TNM Classification of Lung Cancer. J. Thorac. Oncol. 2016, 11, 1204–1223. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, E.; Vrana, K.E. Dopamine beta-hydroxylase and its genetic variants in human health and disease. J. Neurochem. 2020, 152, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Bicker, J.; Alves, G.; Fortuna, A.; Soares-Da-Silva, P.; Falcão, A. In vitro assessment of the interactions of dopamine β-hydroxylase inhibitors with human P-glycoprotein and Breast Cancer Resistance Protein. Eur. J. Pharm. Sci. 2018, 117, 35–40. [Google Scholar] [CrossRef]
- Jen, P.Y.P.; Dixon, J.S. DbetaH-immunoreactive subepithelial nerves in the vas deferens of prostate cancer patients. J. Anat. 2000, 196, 121. [Google Scholar] [CrossRef] [PubMed]
- Tokumoto, M.; Lee, J.-Y.; Fujiwara, Y.; Uchiyama, M.; Satoh, M. Inorganic arsenic induces apoptosis through downregulation of Ube2d genes and p53 accumulation in rat proximal tubular cells. J. Toxicol. Sci. 2013, 38, 815–820. [Google Scholar] [CrossRef]
- Hou, L.; Li, Y.; Wang, Y.; Xu, D.; Cui, H.; Xu, X.; Cong, Y.; Yu, C. UBE2D1 RNA Expression Was an Independent Unfavorable Prognostic Indicator in Lung Adenocarcinoma, but Not in Lung Squamous Cell Carcinoma. Dis. Markers 2018, 2018, 4108919. [Google Scholar] [CrossRef] [PubMed]
- Somwar, R.; Erdjument-Bromage, H.; Larsson, E.; Shum, D.; Lockwood, W.W.; Yang, G.; Sander, C.; Ouerfelli, O.; Tempst, P.J.; Djaballah, H.; et al. Superoxide dismutase 1 (SOD1) is a target for a small molecule identified in a screen for inhibitors of the growth of lung adenocarcinoma cell lines. Proc. Natl. Acad. Sci. USA 2011, 108, 16375–16380. [Google Scholar] [CrossRef]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Doñate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar] [CrossRef]
- Zhan, P.; Shen, X.-K.; Qian, Q.; Zhu, J.-P.; Zhang, Y.; Xie, H.-Y.; Xu, C.-H.; Hao, K.-K.; Hu, W.; Xia, N.; et al. Down-regulation of lysyl oxidase-like 2 (LOXL2) is associated with disease progression in lung adenocarcinomas. Med Oncol. 2012, 29, 648–655. [Google Scholar] [CrossRef]
- Mizuno, K.; Seki, N.; Mataki, H.; Matsushita, R.; Kamikawaji, K.; Kumamoto, T.; Takagi, K.; Goto, Y.; Nishikawa, R.; Kato, M.; et al. Tumor-suppressive microRNA-29 family inhibits cancer cell migration and invasion directly targeting LOXL2 in lung squamous cell carcinoma. Int. J. Oncol. 2016, 48, 450–460. [Google Scholar] [CrossRef]
- Li, F.; Song, Q.-Z.; Zhang, Y.-F.; Wang, X.-R.; Cao, L.-M.; Li, N.; Zhao, L.-X.; Zhang, S.-X.; Zhuang, X.-F. Identifying the EMT-related signature to stratify prognosis and evaluate the tumor microenvironment in lung adenocarcinoma. Front. Genet. 2022, 13, 1008416. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Wei, J. Validation of a Novel Cuproptosis–Related Prognostic Gene Marker and Differential Expression Associated with Lung Adenocarcinoma. Curr. Issues Mol. Biol. 2023, 45, 8502-8518. https://doi.org/10.3390/cimb45100536
Liu T, Wei J. Validation of a Novel Cuproptosis–Related Prognostic Gene Marker and Differential Expression Associated with Lung Adenocarcinoma. Current Issues in Molecular Biology. 2023; 45(10):8502-8518. https://doi.org/10.3390/cimb45100536
Chicago/Turabian StyleLiu, Tingting, and Jianshe Wei. 2023. "Validation of a Novel Cuproptosis–Related Prognostic Gene Marker and Differential Expression Associated with Lung Adenocarcinoma" Current Issues in Molecular Biology 45, no. 10: 8502-8518. https://doi.org/10.3390/cimb45100536
APA StyleLiu, T., & Wei, J. (2023). Validation of a Novel Cuproptosis–Related Prognostic Gene Marker and Differential Expression Associated with Lung Adenocarcinoma. Current Issues in Molecular Biology, 45(10), 8502-8518. https://doi.org/10.3390/cimb45100536