
In Silico Evaluation of Coding and Non-Coding nsSNPs in the Thrombopoietin Receptor (MPL) Proto-Oncogene: Assessing Their Influence on Protein Stability, Structure, and Function
 ,
,
Abstract
:1. Introduction
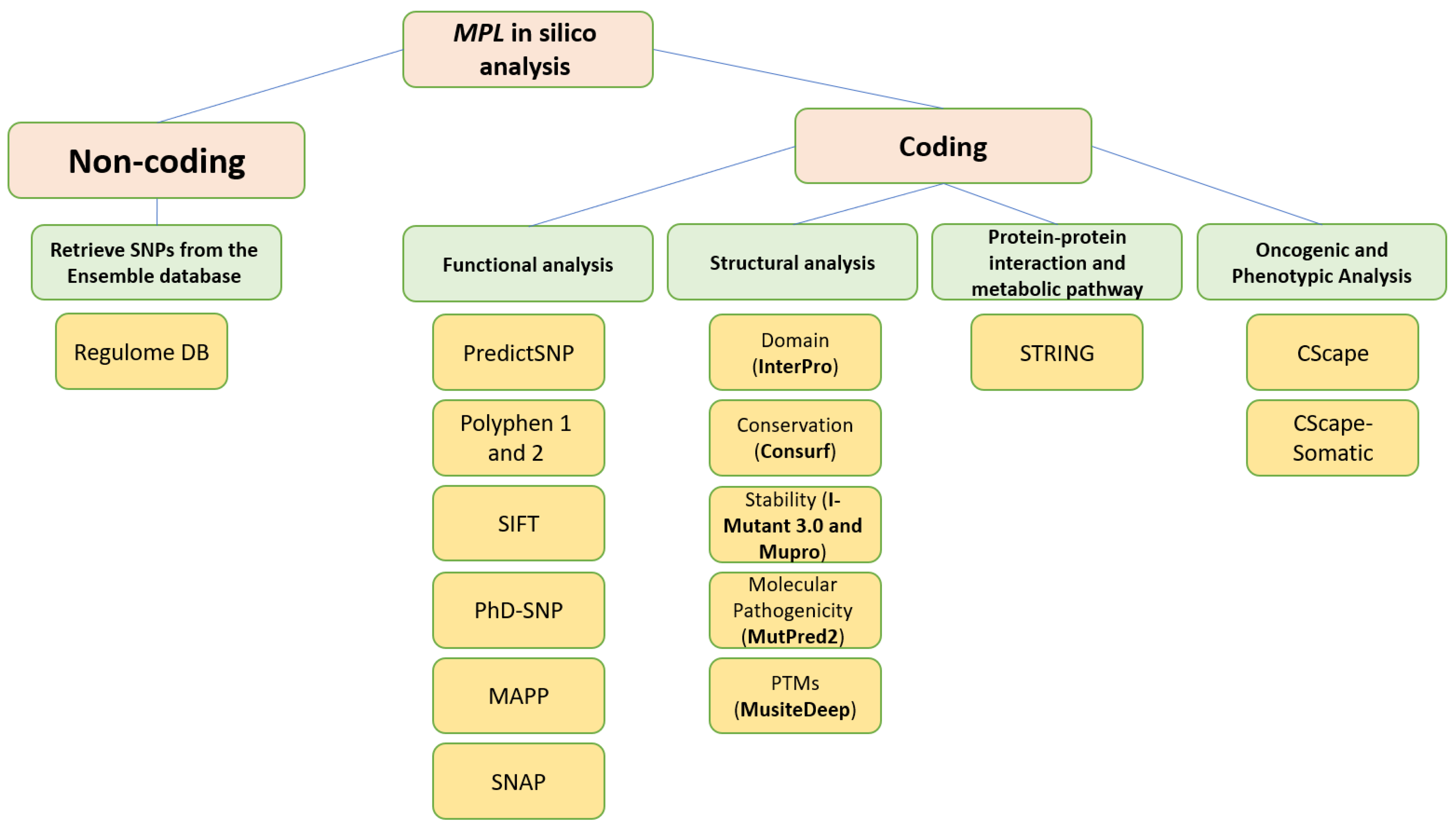
2. Materials and Methods
2.1. Retrieval of nsSNPs from the Database
2.2. Determining the Most Deleterious SNPs
2.3. Identification of nsSNPs within the Domains of the MPL Gene
2.4. Assessment of the Conservation Profile Using ConSurf
2.5. Prediction Alterations in Protein Stability
2.6. Identification of MPL’s Post-Translational Modification Sites Using MusiteDeep
2.7. Predicting Protein–Protein Interactions Using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING)
2.8. Analysis of the Functional Relevance of Non-Coding SNPs (ncSNPs) in the MPL Gene
- Rank 1: strong evidence suggesting potential to affect binding and is associated with the expression of a gene target;
- Rank 2: likely to influence binding;
- Rank 3: lesser likelihood of affecting binding;
- Ranks 4, 5, 6: limited evidence of binding and are typically void of known regulatory functions.
2.9. Assessment of the Structural Impact of nsSNPs on the Human MPL Protein
2.10. Evaluation of Molecular Pathogenicity of nsSNPs
2.11. Oncogenic and Phenotypic Analysis
3. Results
3.1. Identification of Deleterious nsSNPs
3.2. Identification of MPL Gene Domains Using InterPro
3.3. Evolutionary Conservation Analysis
3.4. Protein Stability Prediction
3.5. Identification of MPL Post-Translational Modification Sites
3.6. Determination of MPL Protein Interactions
3.7. Evaluation of the Functional Consequences of Non-Coding SNPs
3.8. Project HOPE Analysis
3.9. Molecular Mechanism of Pathogenicity Prediction
3.10. Oncogenicity Determination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hitchcock, I.S.; Hafer, M.; Sangkhae, V.; Tucker, J.A. The thrombopoietin receptor: Revisiting the master regulator of platelet production. Platelets 2021, 32, 770–778. [Google Scholar] [CrossRef]
- Patel, S.R.; Hartwig, J.H.; Italiano, J.E.J. The biogenesis of platelets from megakaryocyte proplatelets. J. Clin. Investig. 2005, 115, 3348–3354. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.-R.; Chen, J.; Narayanagari, S.-R.; Todorova, T.I.; Aivalioti, M.M.; Ferreira, M.; Ramos, P.M.; Pallaud, C.; Mantzaris, I.; Shastri, A.; et al. Thrombopoietin receptor-independent stimulation of hematopoietic stem cells by eltrombopag. Sci. Transl. Med. 2018, 10, eaas9563. [Google Scholar] [CrossRef] [PubMed]
- Dorsch, M.; Fan, P.-D.; Danial, N.; Rothman, P.; Goff, S. The Thrombopoietin Receptor Can Mediate Proliferation without Activation of the Jak-STAT Pathway. J. Exp. Med. 1997, 186, 1947–1955. [Google Scholar] [CrossRef]
- Hoggatt, J.; Pelus, L.M. Mobilization of hematopoietic stem cells from the bone marrow niche to the blood compartment. Stem Cell Res. Ther. 2011, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.S.; Jiang, S.; Afentoulis, M.; Baumann, C.I.; Schroeder, D.A.; Olson, S.B.; Wong, M.H.; Fleming, W.H. Transplanted adult hematopoietic stems cells differentiate into functional endothelial cells. Blood 2004, 103, 13–19. [Google Scholar] [CrossRef]
- Alev, C.; Nagayama, M.; Asahara, T. Endothelial Progenitor Cells: A Novel Tool for the Therapy of Ischemic Diseases. Antioxid. Redox Signal. 2011, 15, 949–965. [Google Scholar] [CrossRef]
- Bobik, R.; Hong, Y.; Breier, G.; Martin, J.F.; Erusalimsky, J.D. Thrombopoietin stimulates VEGF release from c-Mpl-expressing cell lines and haematopoietic progenitors. FEBS Lett. 1998, 423, 10–14. [Google Scholar] [CrossRef]
- Martínez, C.E.; Smith, P.C.; Palma Alvarado, V.A. The influence of platelet-derived products on angiogenesis and tissue repair: A concise update. Front. Physiol. 2015, 6, 290. [Google Scholar] [CrossRef]
- Kaushansky, K.; Broudy, V.C.; Grossmann, A.; Humes, J.; Lin, N.; Ren, H.P.; Bailey, M.C.; Papayannopoulou, T.; Forstrom, J.W.; Sprugel, K.H. Thrombopoietin expands erythroid progenitors, increases red cell production, and enhances erythroid recovery after myelosuppressive therapy. J. Clin. Investig. 1995, 96, 1683–1687. [Google Scholar] [CrossRef]
- He, X.; Chen, Z.; Jiang, Y.; Qiu, X.; Zhao, X. Different mutations of the human c-mpl gene indicate distinct haematopoietic diseases. J. Hematol. Oncol. 2013, 6, 11. [Google Scholar] [CrossRef]
- Ramensky, V.; Bork, P.; Sunyaev, S. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef] [PubMed]
- Cargill, M.; Altshuler, D.; Ireland, J.; Sklar, P.; Ardlie, K.; Patil, N.; Shaw, N.; Lane, C.R.; Lim, E.P.; Kalyanaraman, N.; et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 1999, 22, 231–238. [Google Scholar] [CrossRef]
- Yue, P.; Moult, J. Identification and analysis of deleterious human SNPs. J. Mol. Biol. 2006, 356, 1263–1274. [Google Scholar] [CrossRef]
- Chatterjee, S.; Pal, J.K. Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol. Cell 2009, 101, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Babeker, M.; Mohamed, A.; Mossalami, G.; Elnasri, H.; Khaier, M.; Eldinelsamani, M. In Silico Analysis of Single Nucleotide Polymorphisms (SNPs) in Human MPL Gene. Int. J. Genet. Genom. 2019, 7, 130–137. [Google Scholar] [CrossRef]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018, bay119. [Google Scholar] [CrossRef]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Birney, E.; Biswas, M.; Bucher, P.; Cerutti, L.; Corpet, F.; Croning, M.D.; et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001, 29, 37–40. [Google Scholar] [CrossRef]
- Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, T.K.; Bateman, A.; Bernard, T.; Binns, D.; Bork, P.; Burge, S.; et al. InterPro in 2011: New developments in the family and domain prediction database. Nucleic Acids Res. 2012, 40, D306–D312. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, D.; Yuchi, J.; He, F.; Jiang, Y.; Cai, S.; Li, J.; Xu, D. MusiteDeep: A deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020, 48, W140–W146. [Google Scholar] [CrossRef]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Venselaar, H.; te Beek, T.A.H.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.; Lin, G.; Nam, H.-J.; Mort, M.; Cooper, D.; Sebat, J.; Iakoucheva, L.; et al. MutPred2: Inferring the molecular and phenotypic impact of amino acid variants. bioRxiv 2017. [Google Scholar] [CrossRef]
- Rogers, M.F.; Shihab, H.A.; Gaunt, T.R.; Campbell, C. CScape: A tool for predicting oncogenic single-point mutations in the cancer genome. Sci. Rep. 2017, 7, 11597. [Google Scholar] [CrossRef]
- Rogers, M.F.; Gaunt, T.R.; Campbell, C. CScape-somatic: Distinguishing driver and passenger point mutations in the cancer genome. Bioinformatics 2020, 36, 3637–3644. [Google Scholar] [CrossRef]
- Plo, I.; Bellanné-Chantelot, C.; Mosca, M.; Mazzi, S.; Marty, C.; Vainchenker, W. Genetic Alterations of the Thrombopoietin/MPL/JAK2 Axis Impacting Megakaryopoiesis. Front. Endocrinol. 2017, 8, 234. [Google Scholar] [CrossRef]
- Hitchcock, I.S.; Kaushansky, K. Thrombopoietin from beginning to end. Br. J. Haematol. 2014, 165, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Germeshausen, M.; Ballmaier, M.; Welte, K. MPL mutations in 23 patients suffering from congenital amegakaryocytic thrombocytopenia: The type of mutation predicts the course of the disease. Hum. Mutat. 2006, 27, 296. [Google Scholar] [CrossRef] [PubMed]
- Deller, M.C.; Kong, L.; Rupp, B. Protein stability: A crystallographer’s perspective. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 72–95. [Google Scholar] [CrossRef]
- Witham, S.; Takano, K.; Schwartz, C.; Alexov, E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins 2011, 79, 2444–2454. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Merlinsky, T.R.; Levine, R.L.; Pronier, E. Unfolding the Role of Calreticulin in Myeloproliferative Neoplasm Pathogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2956–2962. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Baral, A.J.; Beeson, D.; Rivera, J.F.; Ko, A.; Florescu, N.; Birrane, G.; Chen, E.; Mullally, A. Defining the requirements for the pathogenic interaction between mutant calreticulin and MPL in MPN. Blood 2018, 131, 782–786. [Google Scholar] [CrossRef]
- Gadomska, G.; Bartoszewska-Kubiak, A.; Boinska, J.; Matiakowska, K.; Ziołkowska, K.; Haus, O.; Rość, D. Selected Parameters of Angiogenesis and the JAK2, CALR, and MPL Mutations in Patients with Essential Thrombocythemia. Clin. Appl. Thromb. 2018, 24, 1056–1060. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP ID | Missense nsSNPs | PredictSNP | MAPP, PhD-SNP, SIFT, SNAP | PolyPhen1 and 2 |
|---|---|---|---|---|
| rs752112087 | W4R | Deleterious | Deleterious | Damaging |
| rs878854771 | L31P | Deleterious | Deleterious | Damaging |
| rs769423189 | F41L | Deleterious | Deleterious | Damaging |
| rs772445486 | T44I | Deleterious | Deleterious | Damaging |
| rs764333753 | T49A | Deleterious | Deleterious | Damaging |
| rs1361805832 | E100V | Deleterious | Deleterious | Damaging |
| rs1196161699 | F104S | Deleterious | Deleterious | Damaging |
| rs759736065 | L125P | Deleterious | Deleterious | Damaging |
| rs765939441 | G172V | Deleterious | Deleterious | Damaging |
| rs1313333589 | C193Y | Deleterious | Deleterious | Damaging |
| rs1353158125 | L234P | Deleterious | Deleterious | Damaging |
| rs587778519 | G245R | Deleterious | Deleterious | Damaging |
| rs121913611 | R257C | Deleterious | Deleterious | Damaging |
| rs776198457 | D261V | Deleterious | Deleterious | Damaging |
| rs1399857330 | C291R | Deleterious | Deleterious | Damaging |
| rs1171488184 | C291S | Deleterious | Deleterious | Damaging |
| rs919046499 | T293N | Deleterious | Deleterious | Damaging |
| rs113696793 | D295G | Deleterious | Deleterious | Damaging |
| rs1308079365 | P382R | Deleterious | Deleterious | Damaging |
| rs1016116228 | A388D | Deleterious | Deleterious | Damaging |
| rs1006158872 | W435C | Deleterious | Deleterious | Damaging |
| rs121913613 | W491C | Deleterious | Deleterious | Damaging |
| rs775078555 | W491R | Deleterious | Deleterious | Damaging |
| rs1362911656 | L510P | Deleterious | Deleterious | Damaging |
| rs766642690 | Y591D | Deleterious | Deleterious | Damaging |
| rs751884662 | Y626S | Deleterious | Deleterious | Damaging |
| rs753170989 | W632C | Deleterious | Deleterious | Damaging |
| rs536317440 | W632R | Deleterious | Deleterious | Damaging |
| POS | SEQ | Score | Color | b/e | Function |
|---|---|---|---|---|---|
| 4 | W | 0.023 | 5 | b | |
| 31 | L | −0.621 | 7 | b | |
| 41 | F | −0.999 | 9 | b | s |
| 44 | T | −0.648 | 7 | e | |
| 49 | T | −1.072 | 9 | b | s |
| 100 | E | −0.339 | 6 | e | |
| 104 | F | −1.131 | 9 | b | s |
| 125 | L | −0.639 | 7 | b | |
| 172 | G | 0.026 | 5 | b | |
| 193 | C | −0.604 | 7 | b | |
| 234 | L | −0.022 | 5 | b | |
| 245 | G | 0.131 | 5 | e | |
| 257 | R | −1.185 | 9 | e | f |
| 261 | D | −1.186 | 9 | e | f |
| 291 | C | −1.117 | 9 | b | s |
| 293 | T | −1.149 | 9 | e | f |
| 295 | D | −1.03 | 9 | e | f |
| 382 | P | −0.931 | 8 | e | f |
| 388 | A | −0.983 | 9 | b | s |
| 435 | W | −1.013 | 9 | b | s |
| 491 | W | 0.169 | 4 | b | |
| 510 | L | −0.718 | 8 | b | |
| 591 | Y | −0.937 | 8 | b | |
| 626 | Y | −0.937 | 8 | b | |
| 632 | W | −0.77 | 8 | e | f |
| Position | WT | New | SVM2 | RI | DDG Kcal/mol | SVM3 | RI | MUpro | DDG |
|---|---|---|---|---|---|---|---|---|---|
| 31 | L | P | Decrease | 1 | −1.19 | Large decrease | 4 | Decrease | −1.9490598 |
| 41 | F | L | Decrease | 5 | −1.20 | Large decrease | 3 | Decrease | −0.77557062 |
| 44 | T | I | Increase | 2 | −0.20 | Large decrease | 2 | ||
| 49 | T | A | Decrease | 6 | −0.73 | Large decrease | 5 | Decrease | −0.72653759 |
| 100 | E | V | Increase | 1 | 0.08 | Large increase | 2 | ||
| 104 | F | S | Decrease | 9 | −1.85 | Large decrease | 6 | Decrease | −1.6842812 |
| 125 | L | P | Decrease | 5 | −1.78 | Large decrease | 6 | Decrease | −2.4825562 |
| 193 | C | Y | Decrease | 1 | 0.6 | Large decrease | 3 | Decrease | −0.45224808 |
| 257 | R | C | Decrease | 4 | −1.03 | Neutral | 0 | Decrease | −0.50317989 |
| 261 | D | V | Decrease | 1 | 0.00 | Large increase | 1 | Decrease | −0.35365066 |
| 291 | C | S | Decrease | 7 | −0.78 | Large decrease | 3 | Decrease | −0.98271664 |
| 293 | T | N | Decrease | 2 | −0.64 | Neutral | 1 | Decrease | −0.66131688 |
| 295 | D | G | Decrease | 3 | −0.58 | Neutral | 1 | Decrease | −1.4247549 |
| 382 | P | R | Decrease | 7 | −1.08 | Large decrease | 3 | Decrease | −1.0745581 |
| 388 | A | D | Decrease | 0 | −0.4 | Large decrease | 3 | Decrease | −0.81662298 |
| 435 | W | C | Decrease | 8 | −1.48 | Large decrease | 3 | Decrease | −1.0563564 |
| 510 | L | P | Decrease | 2 | −1.12 | Neutral | 0 | Decrease | −2.109342 |
| 591 | Y | D | Decrease | 3 | −0.82 | Large decrease | 3 | Decrease | −1.1085767 |
| 626 | Y | S | Decrease | 6 | −1.28 | Large decrease | 3 | Decrease | −1.2188289 |
| 632 | W | C | Decrease | 8 | −1.37 | Large decrease | 6 | Decrease | −0.76506391 |
| 632 | W | R | Decrease | 7 | −085 | Large decrease | 4 | Decrease | −1.026455 |
| Amino Acid | Position | Post-Translation Modification Site | Score |
|---|---|---|---|
| P | 2 | Hydroxylation | 0.501 |
| T | 180 | Glycosylation | 0.523 |
| S | 228 | Phosphorylation | 0.524 |
| S | 232 | Phosphorylation | 0.551 |
| P | 324 | Hydroxylation | 0.536 |
| K | 339 | Ubiquitination | 0.53 |
| T | 393 | Phosphorylation | 0.542 |
| R | 462 | Methylation | 0.55 |
| Q | 516 | Pyrrolidone carboxylic acid | 0.527 |
| T | 555 | Glycosylation | 0.506 |
| S | 585 | Phosphorylation | 0.553 |
| Chromosome Location | dbSNP IDs | Rank | Score |
|---|---|---|---|
| chr1:43803512..43803513 | rs539244587 | 3a | 0.78994 |
| chr1:43818460..43818461 | rs569647683 | 3a | 0.8738 |
| chr1:43818473..43818474 | rs538671145 | 3a | 0.52742 |
| chr1:43818553..43818554 | rs186807040 | 2b | 0.82679 |
| chr1:43818558..43818559 | rs554711682 | 2b | 0.8288 |
| chr1:43818713..43818714 | rs144331455 | 2b | 0.8153 |
| chr1:43819939..43819940 | rs570264040 | 3a | 0.85638 |
| chr1:43820116..43820117 | rs536844021 | 3a | 0.73862 |
| Residue | AA Changes | Structure | AA Properties |
|---|---|---|---|
| L31P | rs878854771 |  |
|
| F41L | rs769423189 |  |
|
| T49A | rs764333753 |  |
|
| F104S | rs1196161699 |  |
|
| L125P | rs759736065 |  |
|
| C193Y | rs1313333589 |  |
|
| C291R | rs1399857330 |  |
|
| P382R | rs1308079365 |  |
|
| A388D | rs1016116228 |  |
|
| W435C | rs1006158872 |  |
|
| Y591D | rs766642690 |  |
|
| Y626S | rs751884662 |  |
|
| W632C | rs753170989 |  |
|
| AA Variation | MutPred2 Score | Molecular Mechanism with p Value Less than 0.05 |
|---|---|---|
| L31P | 0.265 | - |
| F41L | 0.706 | Altered Transmembrane protein Loss of Strand Gain of Disulfide linkage at C40 |
| T44I | 0.458 | - |
| T49A | 0.752 | Altered Metal binding Loss of Strand Altered Transmembrane protein Gain of Disulfide linkage at C50 |
| E100V | 0.481 | - |
| F104S | 0.922 | Altered Ordered interface Altered Metal binding Altered Transmembrane protein Loss of Loop Altered Stability |
| L125P | 0.811 | Altered Stability Altered Transmembrane protein Gain of ADP-ribosylation at R120 Loss of Proteolytic cleavage at D128 |
| C193Y | 0.562 | Loss of Intrinsic disorder Gain of Strand Gain of Loop Altered Transmembrane protein Gain of O-linked glycosylation at T192 |
| R257C | 0.750 | Altered Transmembrane protein |
| D261V | 0.725 | Loss of Loop Altered Transmembrane protein |
| C291S | 0.915 | Loss of Disulfide linkage at C291 Altered Transmembrane protein |
| T293N | 0.742 | Altered Transmembrane protein Gain of Disulfide linkage at C291 Loss of N-linked glycosylation at N298 |
| D295G | 0.808 | Altered Transmembrane protein Loss of Disulfide linkage at C291 Loss of N-linked glycosylation at N298 |
| P382R | 0.779 | Gain of Strand Altered Transmembrane protein |
| A388D | 0.525 | Altered Transmembrane protein |
| W435C | 0.790 | Altered Metal binding Altered Transmembrane protein Loss of Strand Gain of Disulfide linkage at W435 |
| L510P | 0.735 | - |
| Y591D | 0.318 | - |
| Y626S | 0.324 | - |
| W632C | 0.464 | - |
| W632R | 0.444 | - |
| CScape | CScape-Somatic | ||||||
|---|---|---|---|---|---|---|---|
| Variant ID | SNP | Input and Assembly GRCh38 | Coding Score | Message | Input and Assembly GRCh37 | Non-Coding Score | Message |
| rs769423189 | F41L | 1,43338140,T,C | 0.596320 | Oncogenic | 1,43338140,T,C | - | - |
| rs764333753 | T49A | 1,43338164,A,G | 0.626069 | Oncogenic | 1,43338164,A,G | - | - |
| rs1196161699 | F104S | 1,43338640,T,C | 0.586258 | Oncogenic | 1,43338640,T,C | - | - |
| rs759736065 | L125P | 1,43338703,T,C | 0.872560 | Oncogenic | 1,43338703,T,C | - | - |
| rs121913611 | R257C | 1,43340042,C,T | 0.745871 | Oncogenic | 1,43340042,C,T | - | - |
| rs776198457 | D261V | 1,43340055,A,T | 0.765347 | Oncogenic | 1,43340055,A,T | - | - |
| rs1171488184 | C291S | 1,43340405,G,C | 0.714650 | Oncogenic | 1,43340405,G,C | 0.455926 | Passenger |
| rs919046499 | T293N | 1,43340411,C,A | 0.594031 | Oncogenic | 1,43340411,C,A | 0.223170 | Passenger |
| rs113696793 | D295G | 1,43340417,A,G | 0.869262 | Oncogenic | 1,43340417,A,G | 0.256742 | Passenger |
| rs1308079365 | P382R | 1,43346609,C,G | 0.726814 | Oncogenic | 1,43346609,C,G | - | - |
| rs1016116228 | A388D | 1,43346627,C,A | 0.643915 | Oncogenic | 1,43346627,C,A | - | - |
| rs1006158872 | W435C | 1,43346931,G,C | 0.853135 | Oncogenic | 1,43346931,G,C | 0.423081 | Passenger |
| rs1362911656 | L510P | 1,43349323,T,C | 0.701219 | Oncogenic | 1,43349323,T,C | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-nakhle, H.H.; Yagoub, H.S.; Anbarkhan, S.H.; Alamri, G.A.; Alsubaie, N.M. In Silico Evaluation of Coding and Non-Coding nsSNPs in the Thrombopoietin Receptor (MPL) Proto-Oncogene: Assessing Their Influence on Protein Stability, Structure, and Function. Curr. Issues Mol. Biol. 2023, 45, 9390-9412. https://doi.org/10.3390/cimb45120589
Al-nakhle HH, Yagoub HS, Anbarkhan SH, Alamri GA, Alsubaie NM. In Silico Evaluation of Coding and Non-Coding nsSNPs in the Thrombopoietin Receptor (MPL) Proto-Oncogene: Assessing Their Influence on Protein Stability, Structure, and Function. Current Issues in Molecular Biology. 2023; 45(12):9390-9412. https://doi.org/10.3390/cimb45120589
Chicago/Turabian StyleAl-nakhle, Hakeemah H., Hind S. Yagoub, Sadin H. Anbarkhan, Ghadah A. Alamri, and Norah M. Alsubaie. 2023. "In Silico Evaluation of Coding and Non-Coding nsSNPs in the Thrombopoietin Receptor (MPL) Proto-Oncogene: Assessing Their Influence on Protein Stability, Structure, and Function" Current Issues in Molecular Biology 45, no. 12: 9390-9412. https://doi.org/10.3390/cimb45120589