

Alzheimer’s Disease beyond Calcium Dysregulation: The Complex Interplay between Calmodulin, Calmodulin-Binding Proteins and Amyloid Beta from Disease Onset through Progression

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
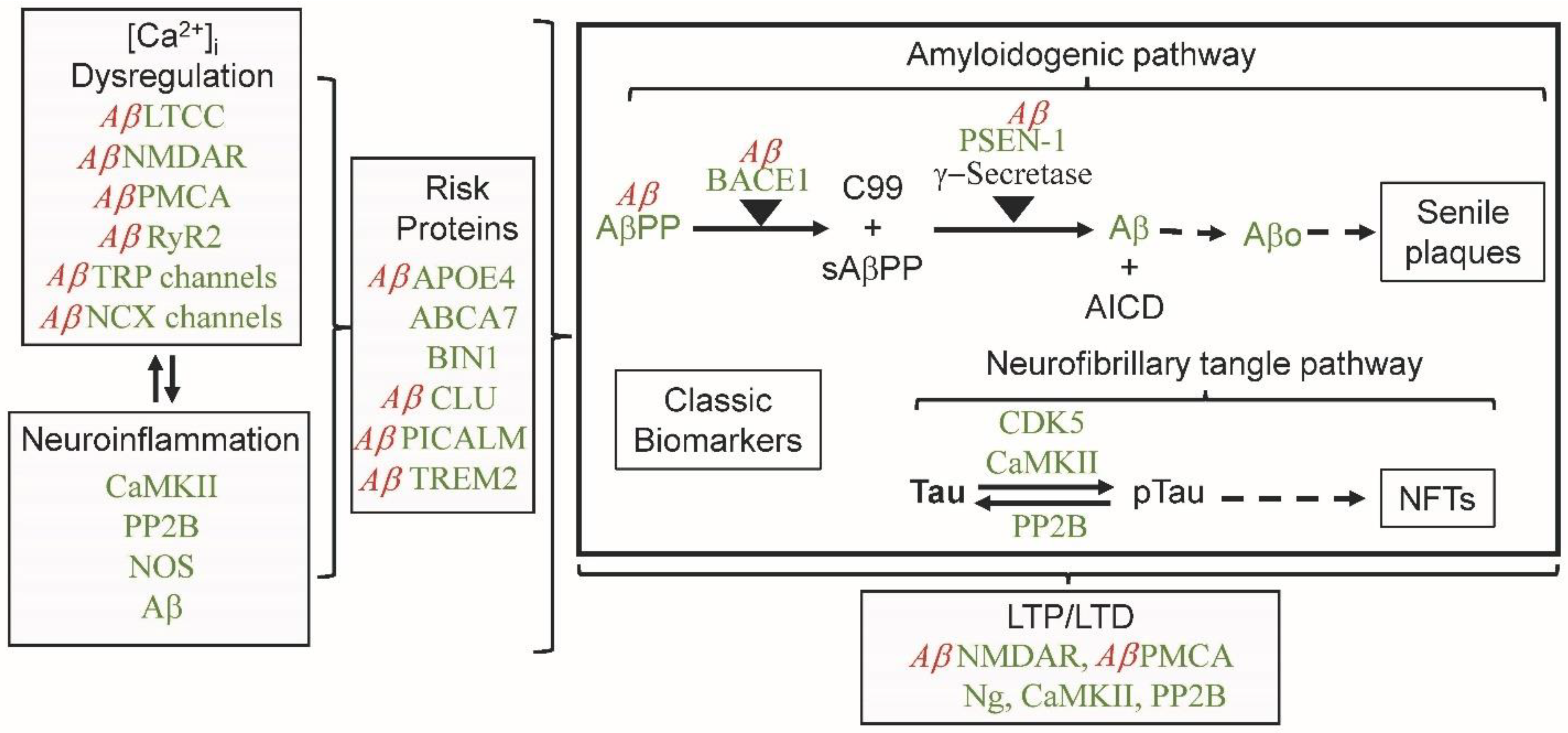
2. Calcium Dysregulation and Calmodulin
2.1. Calcium Regulation of Calmodulin
2.2. Calmodulin Hypothesis of Alzheimer’s Disease
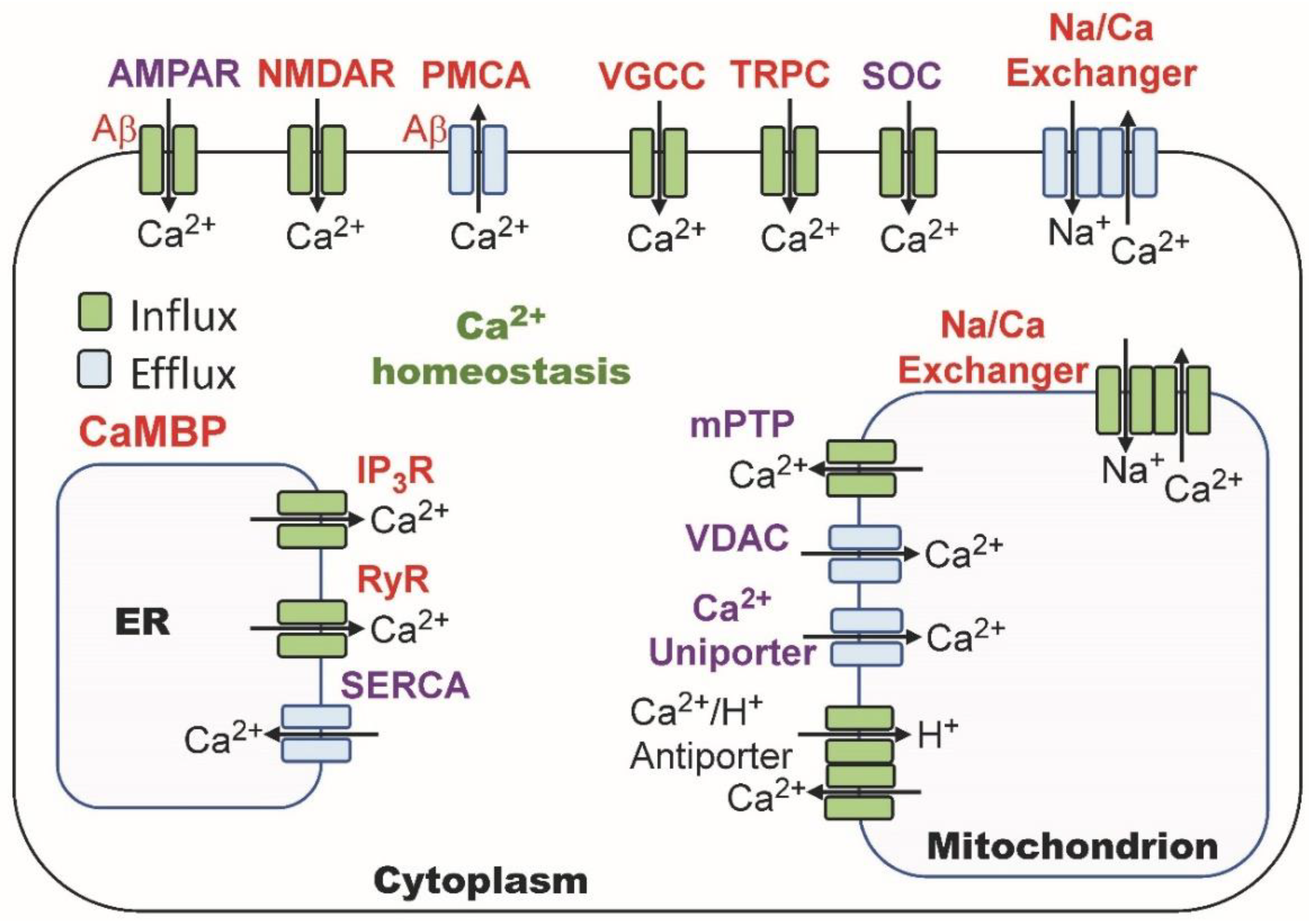
2.3. Calmodulin Regulation of Calcium Homeostasis
3. CaMBPs and Neuroinflammation
4. CaM, CaMBPs and Aβ in Amyloidogenesis
BACE1 Regulation by CaM and Aβ
5. Tau Phosphorylation
6. CaM, Aβ, LTP and LTD
6.1. NMDAR: A CaM-Binding Ion Channel
6.2. AMPAR: Ion Channel Regulated by CaMBPs
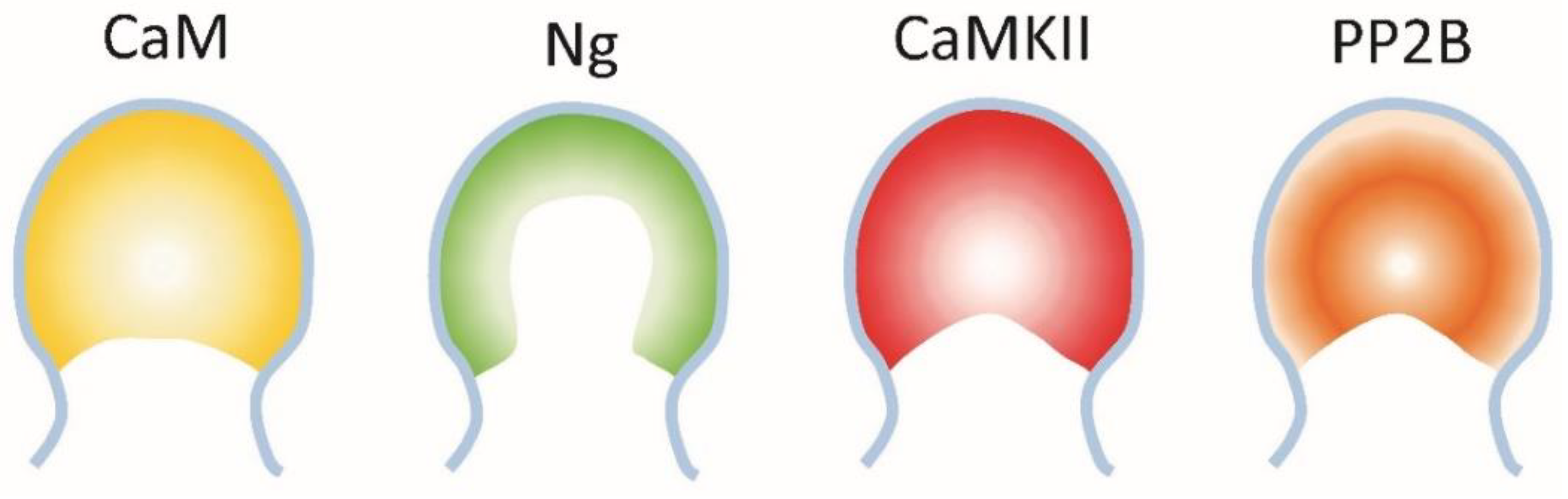
6.3. The Interaction between Neurogranin, CaMKII and PP2B
7. Calmodulin and Synaptic Vesicle Exocytosis
8. Final Comments
Funding
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. Alzheimer’s Association Report. 2023 Alzheimer’s Facts and Figures; Alzheimer’s Association: Chicago, IL, USA, 2023; Volume 19, pp. 1598–1695. [Google Scholar]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Cummings, J.; Zhou, Y.; Lee, G.; Zhong, K.; Fonseca, J.; Cheng, F. Alzheimer’s disease drug development pipeline: 2023. Transl. Res. Clin. Intervent. 2023, 7, e12385. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Greenberg, M.E. Calcium signaling in neurons: Molecular mechanisms and cellular consequences. Science 1995, 268, 239–247. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Carafoli, E. Intracellular calcium homeostasis. Ann. Rev. Biochem. 1987, 56, 395–433. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Cecchi, C. Calcium dyshomeostasis in Alzheimer’s disease pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann. N. Y. Acad. Sci. 1994, 747, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. The role of calcium in brain aging: Reexamination of a hypothesis. Aging 1989, 1, 17–34. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Guan, P.-P.; Cao, L.-L.; Wang, P. Elevating the levels of calcium ions exacerbate Alzheimer’s disease via inducing the production and aggregation of b-amyloid protein and phosphorylated tau. Int. J. Mol. Sci. 2021, 22, 5900. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Green, K.N.; LaFerla, F.M. Linking calcium to Abeta and Alzheimer’s disease. Neuron 2008, 59, 190–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Webber, E.K.; Fivaz, M.; Stutzmann, G.E.; Griffoen, G. Cytosolic calcium: Judge jury and executioner of neurodegeneration in Alzheimer’s disease and beyond. Alzheimer’s Dement. 2023, 1–17. [Google Scholar] [CrossRef]
- Sharma, R.K.; Parameswaran, S. Calmodulin-binding proteins: A journey of 40 years. Cell Calcium 2018, 75, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Biber, A.; Schmid, G.; Hempel, K. Calmodulin content in specific brain areas. Exp. Brain Res. 1984, 56, 323–326. [Google Scholar] [CrossRef]
- McLachlan, D.R.C.; Wong, L.; Bergeron, C.; Baimbridge, K.G. Calmodulin and calbindin D28K in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1987, 1, 171–179. [Google Scholar] [CrossRef]
- Rhoads, A.R.; Friedberg, F. Sequence motifs for calmodulin recognition. FASEB J. 1997, 11, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell. Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013, 280, 5551–5565. [Google Scholar] [CrossRef]
- Grant, B.M.M.; Enomoto, M.; Ikura, M.; Marshall, C.B. A non-canonical calmodulin target motif comprising a polybasic region and lapidates terminal residue regulates localization. Int. J. Mol. Sci. 2020, 21, 2751. [Google Scholar] [CrossRef] [Green Version]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin binding proteins and Alzheimer’s disease: A review. J. Alzheimer’s Dis. 2015, 46, 553–569. [Google Scholar] [CrossRef] [Green Version]
- O’Day, D.H.; Huber, R.J. Calmodulin binding proteins and neuroinflammation in multiple neurodegenerative diseases. BMC Neurosci. 2022, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H.; Myre, M.A. Calmodulin-binding domains in Alzheimer’s disease proteins: Extending the calcium hypothesis. Biochem. Biophys. Res. Commun. 2004, 230, 1051–1054. [Google Scholar] [CrossRef]
- Berrocal, M.; Sepulveda, M.R.; Vazquez-Hernandez, M.; Mata, A.M. Calmodulin antagonizes amyloid-βpeptides-mediated inhibition of brain plasma membrane Ca(2+)-ATPase. Biochim. Biophys. Acta 2012, 1822, 961–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbacho, I.; Berrocal, M.; Török, K.; Mata, A.M.; Gutierrez-Merino, C. High affinity binding of amyloid β-peptide to calmodulin: Structural and functional implications. Biochem. Biophys. Res. Commun. 2017, 486, 992–997. [Google Scholar] [CrossRef] [Green Version]
- O’Day, D.H. Calmodulin binding proteins and Alzheimer’s disease: Biomarkers, regulatory enzymes and receptors that are regulated by calmodulin. Int. J. Mol. Sci. 2020, 21, 7344. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H. Calmodulin binding domains in critical risk proteins involved in neurodegeneration. Curr. Issues Mol. Biol. 2022, 44, 5802–5814. [Google Scholar] [CrossRef]
- Casal, C.; Tusell, J.M.; Serratosa, J. Role of calmodulin in the differentiation/activation of microglial cells. Brain Res. 2001, 902, 101–107. [Google Scholar] [CrossRef]
- Catterall, W.A.; Few, A.P. Calcium channel regulation and presynaptic plasticity. Neuron 2008, 59, 882–901. [Google Scholar] [CrossRef] [Green Version]
- Blundon, J.A.; Zakharenko, S.S. Dissecting the components of long-term potentiation. Neuroscientist 2008, 14, 598–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhof, T.C. The synaptic vesicle cycle. Ann. Rev. Neurosci. 2004, 27, 509–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.C.; Wolff, J. Calmodulin binds to both microtubule-associated protein 2 and tau proteins. J. Biol. Chem. 1984, 259, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Chemin, J.; Taiakina, V.; Monteil, A.; Piazza, M.; Guan, W.; Stephens, R.F.; Kitmitto, A.; Pang, Z.P.; Dolphin, A.C.; Perez-Reyes, E. Calmodulin regulates Cav3 T-type channels at their gating brake. J. Biol. Chem. 2017, 292, P20010–P20031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Vogel, H.J. Structural basis for the regulation of L-type voltage-gated calcium channels: Interactions between the N-terminal cytoplasmic domain and Ca2+-calmodulin. Front. Mol. Neurosci. 2012, 12, 38. [Google Scholar] [CrossRef] [Green Version]
- Iacobucci, G.J.; Popescu, G.K. Resident calmodulin primes NMDA receptors for Ca2+-dependent inactivation. Biophys. J. 2017, 113, 2236–2248. [Google Scholar] [CrossRef] [Green Version]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Kushnireva, L.; Korkotian, E.; Segal, M. Calcium Sensors STIM1 and STIM2 Regulate Different Calcium Functions in Cultured Hippocampal Neurons. Front. Synaptic Neurosci. 2021, 12, 573714. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Jardin, I.; Salido, G.M.; Rosado, J.A. Role of STIM2 in cell function and physiopathology. J. Physiol. 2017, 595, 3111–3128. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Capacitative calcium entry. Biochem. J. 1995, 312, 1–11. [Google Scholar] [CrossRef]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, H.; Liu, J.; Popugaeva, E.; Xu, N.J.; Feske, S.; White, C.L., 3rd; Bezprozvanny, I. Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 2014, 82, 79–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wu, G.; Yang, Y.; Fu, S.; Liu, X.; Kang, H.; Yang, X.; Su, X.-C.; Shen, Y. Calmodulin dissociates the SITM1-Orai complex and STIM1 oligomers. Nature Commun. 2018, 8, 1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.; Hofmann, T.; Montell, C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol. Cell. 2007, 25, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; He, Q.; Wang, J. TRPC channels and Alzheimer’s disease. Adv. Exp. Med. Biol. 2017, 976, 73–83. [Google Scholar]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common reature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path toward precision medicine for neuroinflammatory mechanisms in Alzheimer’s disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Behnke, V.; Langmann, T. Neuroinflammation in neuronal ceroid lipofuscinosis. Ophthalmologe 2021, 118, 98–105. [Google Scholar] [CrossRef]
- Bohush, A.; Leśniak, W.; Weis, S.; Filipek, A. Calmodulin and its binding proteins in Parkinson’s disease. Int. J. Mol. Med. 2021, 22, 2016. [Google Scholar] [CrossRef] [PubMed]
- Bright, F.; Werry, E.L.; Dobson-Stone, C.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kassiou, M.; et al. Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol. 2019, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.J.M.; Kwakowsky, A. The role of microglia and astrocytes in Huntington’s disease. Front. Mol. Neurol. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer brain. J. Neurochem. 2020, 154, 583–587. [Google Scholar] [CrossRef]
- Phillips, E.C.; Croft, C.L.; Kurbatskaya, K.; O’Neill, M.J.; Hutton, M.L.; Hanger, D.P.; Garwood, C.J.; Nobile, W. Astrocytes and neuroinflammation in Alzheimer’s disease. Biochem. Soc. Trans. 2014, 42, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Stull, J.G. Calmodulin-dependent regulation of inducible and neuronal nitric-oxide synthase. J. Biol. Chem. 1998, 273, 27430–27437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venturini, G.; Colasanti, M.; Persichini, T.; Fioravanti, E.; Ascenzi, P.; Palomba, L.; Cantoni, O.; Musci, G. Beta-amyloid inhibits NOS activity by subtracting NADPH availability. FASEB J. 2002, 16, 1970–1972. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-b receptors: The good, the bad and the prion protein. J. Biol. Chem. 2016, 291, 3174–3183. [Google Scholar] [CrossRef] [Green Version]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of amyloid β oligomers (AβOs) in Alzheimer’s disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Kelly, J.F.; Aggarwal, N.T.; Shah, R.C.; Wilson, R.S. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006, 66, 1837–1844. [Google Scholar] [CrossRef]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Khezri, M.R.; et al. Therapeutic potential of ADAM10 modulation in Alzheimer’s disease: A review of the current evidence. Cell Commun. Signal. 2023, 21, 60. [Google Scholar]
- Chen, P.B.; Kawaguchi, R.; Blum, C.; Achiro, J.M.; Coppola, G.; O’Dell, T.J.; Martin, K.C. Mapping gene expression in excitatory neurons during hippocampal late-phase long-term potentiation. Front. Mol. Neurosci. 2017, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Brit. J. Pharm. 2019, 176, 3489–3507. [Google Scholar] [CrossRef]
- Poejo, J.; Salazar, J.; Mata, A.M.; Gutierrez-Merino, C. The relevance of amyloid b-calmodulin complexation in neurons and brain degeneration in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 4976. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H. Calmodulin and amyloid beta as coregulators of critical events during the onset and progression of Alzheimer’s disease. Int. J. Mol. Sci. 2023, 24, 1393–1401. [Google Scholar] [CrossRef]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and neurodegenerative diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef]
- Canobbio, I.; Catricalà, S.; Balduini, C.; Torti, M. Calmodulin regulates the non-amyloidogenic metabolism of amyloid precursor protein in platelets. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 500–506. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, C.; Pei, G. α-secretase ADAM10 physically interacts with b-secretase BACE1 in neurons and regulates proteolysis. J. Mol. Cell Biol. 2018, 10, 411–422. [Google Scholar] [CrossRef]
- Chavez, S.E.; O’Day, D.H. Calmodulin binds to and regulates the activity of beta-secretase (BACE1). In Current Research on Alzheimer’s Disease; Nova Science Publishers, Inc.: Hauppage, NY, USA, 2007; Volume 1, pp. 37–47. [Google Scholar]
- Manzine, P.R.; Ettchero, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olioquegui, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Bomed. Phamacotherapy 2019, 113, 108661. [Google Scholar] [CrossRef]
- O’Day, D.H. Phytochemical Interactions with Calmodulin and Critical Calmodulin Binding Proteins Involved in Amyloidogenesis in Alzheimer’s Disease. Biomolecules 2023, 13, 678. [Google Scholar] [CrossRef]
- Giliberto, L.; Borghi, R.; Piccini, A.; Mangerini, R.; Sorbi, S.; Cirmena, G.; Garuti, A.; Ghetti, B.; Tagliavini, F.; Mughal, M.R.; et al. Mutant presenilin 1 increases the expression and activity of BACE1. J. Biol. Chem. 2009, 284, 9027–9038. [Google Scholar] [CrossRef] [Green Version]
- Maloney, B.; Lahiri, D.K. The Alzheimer’s amyloid b-peptide (Ab) binds a specific DNA Aβ-interacting domain (AβID) in the APP, BACE1 and APOE promoters in a sequence specific manner: Characterizing a new regulatory motif. Gene 2011, 488, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogel, H.; Frere, S.; Segev, O.; Bharill, S.; Shapira, I.; Gazit, N.; O’Malley, T.; Slomowitz, E.; Berdichevsky, Y.; Walsh, D.M.; et al. APP homodimers transduce an amyloid-β mediated increase in release probability at excitatory synapses. Cell Rep. 2014, 17, 1560–1576. [Google Scholar] [CrossRef] [Green Version]
- Ohki, Y.; Shimada, N.; Tominaga, A.; Osawa, S.; Higo, T.; Yokoshima, S.; Fukuyama, T.; Tomita, T.; Iwatsubo, T. Binding of longer Aβ to transmembrane domain 1 of presenilin 1 impacts on Aβ42 generation. Mol. Neurodegen. 2014, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Michno, K.; Knight, D.; Campusano, J.M.; van de Hoef, D.; Boulianne, G.L. Intracellular calcium deficits in Drosophila cholinergic neurons expressing wild type or FAD-mutant presenilin. PLoS ONE 2009, 4, e6904. [Google Scholar] [CrossRef]
- Padilla, R.; Maccioni, R.B.; Avila, J. Calmodulin binds to a tubulin binding site of the microtubule-associated protein tau. Mol. Cell Biochem. 1990, 97, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Geise, K.P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 2015, 8, 78. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.J.; Catalano, A.; O’Day, D.H. Cyclin-dependent kinase 5 is a calmodulin-binding protein that associates with puromycin-sensitive aminopeptidase in the nucleus of Dictyostelium. Biochem. Biophys. Acta 2013, 1833, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Stefan, M.I.; Edelstein, S.J.; Le Novere, N. An allosteric model of calmodulin explains differential activation of PP2B and CaMKII. Proc. Natl. Acad. Sci. USA 2008, 105, 10768–10773. [Google Scholar] [CrossRef]
- Yu, D.Y.; Tong, L.; Song, G.J.; Lin, W.L.; Zhang, L.Q.; Bai, W.; Gong, H.; Yin, Y.X.; Wei, Q. Tau binds both subunits of calcineurin, and binding is impaired by calmodulin. Biochem. Biophys. Acta 2008, 1783, 2255–2261. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y. Molecular mechanism of hippocampal long-term potentiation—Towards multiscale understanding of learning and memory. Neurosci. Res. 2022, 175, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Alonso, J.; Nicoll, R.A. AMPA receptor trafficking and LTP: Carboxy-termini, amino-termini and TARPS. Neuropharm. 2021, 197, 108710. [Google Scholar] [CrossRef] [PubMed]
- Sumi, T.; Harada, K. Mechanism underlying hippocampal long-term potentiation and depression based on competition between endocytosis and exocytosis of AMPA receptors. Sci. Rep. 2020, 10, 14711. [Google Scholar] [CrossRef] [PubMed]
- Stacho, M.; Manahan-Vaughan, D. The intriguing contribution of hippocampal long-term depression to spatial learning and long-term memory. Front. Behav. Neurosci. 2020, 16, 806356. [Google Scholar] [CrossRef] [PubMed]
- D’Alcantara, P.; Schiffmann, S.N.; Swillens, S. Bidirectional synaptic plasticity as a consequence of interdependent Ca2+-controlled phosphorylation and dephosphorylation pathways. Eur. J. Neurosci. 2003, 17, 2521–2528. [Google Scholar] [CrossRef]
- Li, L.; Massimo, L.; Cole, S.; Novere, N.L.; Edelstein, S.J. Neurogranin stimulates Ca2+/calmodulin-dependent kinase II by suppressing calcineurin activity at specific calcium spike frequencies. PLoS Comput. Biol. 2020, 16, e1006991. [Google Scholar] [CrossRef] [Green Version]
- Rostas, J.A.P.; Skelding, K.A. Calcium/calmodulin-stimulated protein kinase II (CaMKII): Different functional outcomes from activation, depending on the cellular microenvironment. Cells 2023, 12, 401. [Google Scholar] [CrossRef]
- Creamer, T.P. Calcineurin. Cell Commun. Signal. 2020, 18, 137. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- Ehlers, M.D.; Zhang, S.; Bernhardt, J.P.; Huganir, R.L. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell 1996, 84, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [PubMed] [Green Version]
- Sang, L.; Vieira, D.C.O.; Yue, D.T.; Ben-Johny, M.; Dick, I.E. The molecular basis of the inhibition of CaV1 calcium-dependent inactivation by the distal carboxy tail. J. Biol. Chem. 2021, 296, 100502. [Google Scholar] [CrossRef] [PubMed]
- Ataman, Z.A.; Gakhar, L.; Sorenson, B.R.; Hell, J.W.; Shea, M.A. The NMDA receptor NR1 C1 region bound to calmodulin: Structural insights into functional differences between homologous domains. Structure 2007, 15, 1603–1617. [Google Scholar]
- Bej, A.; Ames, J.B. Chemical shift assignments of calmodulin bound to the GluN1 C0 domain (residues 841-865) of the NMDA receptor. Biomol. NMR Assign. 2023, 17, 61–65. [Google Scholar] [CrossRef]
- Zhong, L.; Gerges, N.Z. Neurogranin regulates metaplasticity. Front. Mol. Neurosci. 2020, 12, 2019. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, L.; Chandrasekar, A.; Wand, X.; Putkey, J.A.; Waxham, M.N. Neurogranin alters the structure and calmodulin binding properties of calmodulin. J. Biol. Chem. 2014, 3, 14644–14655. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.; Kessels, H.W.; Alfonso, S.; Aow, J.; Fox, R.; Malinow, R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4027–4032. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.W.; Klyubin, I.; Anwyl, R.; Rowan, M.J. GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20504–20509. [Google Scholar] [CrossRef] [PubMed]
- Malinow, R. New developments on the role of NMDA receptors in Alzheimer’s disease. Curr. Opin. Neurobiol. 2012, 22, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulme, S.R.; Jones, O.D.; Abraham, W.C. Emerging roles of metaplasticity in behaviour and disease. Trends Neurosci. 2013, 36, 353–362. [Google Scholar] [CrossRef]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Cherry, T.; Bies, C.E.; Florence, M.A.; Gerges, N.Z. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009, 28, 3027–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migues, P.V.; Hardt, O.; Wu, D.C.; Gamache, K.; Sacktor, T.C.; Wang, Y.T.; Nader, K. PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat. Neurosci. 2010, 13, 630–634. [Google Scholar] [CrossRef]
- Lee, H.K.; Barbarosie, M.; Kameyama, K.; Bear, M.F.; Huganir, R.L. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 2000, 405, 955–959. [Google Scholar] [CrossRef]
- Purkey, A.M.; Dell’Acqua, M.L. Phosphorylation-dependent regulation of Ca2+-permeable AMPA receptors during hippocampal synaptic plasticity. Front. Synaptic Neurosci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Schwenk, J.; Fakler, B. Building of AMPA-type glutamate receptors in the endoplasmic reticulum and its implication for excitatory neurotransmission. J. Physiol. 2021, 599, 2639–2653. [Google Scholar] [CrossRef]
- Chowdhury, D.; Hell, J.W. Ca2+/calmodulin binding to PSD-95 downregulates Its palmitoylation and AMPARs in long-term depression. Front. Synaptic Neurosci. 2019, 11, 6. [Google Scholar] [CrossRef]
- Won, S.; Levy, J.M.; Nicoll, R.A.; Roche, K.W. MAGUKs: Multifaceted synaptic organizers. Curr. Opin. Neurobiol. 2017, 43, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dore, K.; Carrico, Z.; Alfonso, S.; Marino, M.; Koymans, K.; Kessels, H.W.; Malinow, R. PSD-95 protects synapses from b-amyloid. Cell Rep. 2021, 35, 10914. [Google Scholar] [CrossRef]
- Opazo, P.; Labrecque, S.; Tigaret, C.M.; Frouin, A.; Wiseman, P.W.; De Koninck, P.; Choquet, D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron 2010, 67, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Bissen, D.; Foss, F.; Acker-Palmer, A. AMPA receptors and their minions: Auxiliary proteins in AMPA receptor trafficking. Cell. Mol. Life Sci. 2019, 76, 2133–2169. [Google Scholar]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Guntupalli, S.; Jang, S.E.; Zhu, T.; Huganir, R.L.; Widagdo, J.; Anggono, V. GluA1 subunit ubiquitination mediates amyloid-β-induced loss of surface α-amino-3- hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. J. Biol. Chem. 2017, 292, 8186–8194. [Google Scholar] [CrossRef] [Green Version]
- Pak, J.H.; Huang, F.L.; Li, J.; Huang, K.-P. Involvement of neurogranin in the modulation of calcium/calmodulin-dependent protein kinase II, synaptic plasticity, and spatial learning: A study with knockout mice. Proc. Natl Acad. Sci. USA 2000, 97, 11232–11237. [Google Scholar] [CrossRef]
- Carlyle, B.C.; Kandigian, S.E.; Kreuzer, J.; Das, S.; Trombetta, B.A.; Kuo, Y.; Bennett, D.A.; Schneider, J.A.; Petyuk, V.A.; Kitchen, R.R.; et al. Synaptic proteins associated with cognitive performance and neuropathology in older humans revealed by multiplexed fractionated proteomics. Neurobiol. Aging 2021, 105, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.H.; Baxter, M.G. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nature Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Reese, L.C.; Laezza, F.; Woltjer, R.; Taglialatela, G. Dysregulated phosphorylation of Ca(2+)/calmodulin-dependent protein kinase II-alpha in the hippocampus of subjects with mild cognitive impairment and Alzheimer’s disease. J. Neurochem. 2011, 119, 791–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, S.E.; Li, Y.; Todd, K.W.; Herries, E.M.; Henson, R.L.; Gray, J.D.; Wang, G.; Graham, D.L.; Shaw, L.M.; Trojanowski, J.Q.; et al. Emerging cerebrospinal fluid biomarkers in autosomal dominant Alzheimer’s disease. Alzheimer’s Dement. 2019, 15, 655–665. [Google Scholar] [CrossRef]
- Sutphen, C.L.; McCue, L.; Herries, E.M.; Xiong, C.; Ladenson, J.H.; Holtzman, D.M.; Fagan, A.M.; ADNI. Longitudinal decreases in multiple cerebrospinal fluid biomarkers of neuronal injury in symptomatic late onset Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Davidsson, P.; Blennow, K. Neurochemical dissection of synaptic pathology in Alzheimer’s disease. Int. Psychogeriatr. 1998, 10, 11–23. [Google Scholar] [CrossRef]
- Thorsell, A.; Bjerke, M.; Gobom, J.; Brunhage, E.; Vanmechelen, E.; Andreasen, N.; Hansson, O.; Minthon, L.; Zetterberg, H.; Blennow, K. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res. 2010, 1362, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y. Long-term potentiation: Two pathways meet at neurogranin. EMBO J. 2009, 28, 2859–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanovic, N.; Davidsson, P.; Gottfries, J.; Volkna, I.; Winblad, B.; Blennow, K. Regional and cellular distribution of synaptic proteins in the normal human brain. Brain Aging 2002, 2, 18–30. [Google Scholar]
- Huang, K.P.; Huang, F.L. Calcium-sensitive translocation of calmodulin and neurogranin between Soma and dendrites of mouse hippocampal CA1 neurons. ACS Chem. Neurosci. 2011, 2, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Petersen, A.; Gerges, N.Z. Neurogranin regulates CaM dynamics at dendritic spines. Sci. Rep. 2015, 5, 11135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martzen, M.R.; Slemmon, J.R. The dendritic peptide neurogranin can regulate a calmodulin-dependent target. J. Neurochem. 1995, 64, 92–100. [Google Scholar] [CrossRef]
- Liu, W.; Lin, H.; He, X.; Chen, L.; Dai, Y.; Jia, W.; Xue, X.; Tau, J.; Chen, L. Neurogranin as a cognitive biomarker in cerebrospinal fluid and blood exosomes for Alzheimer’s disease and mild cognitive impairment. Translat. Psych. 2020, 10, 125. [Google Scholar] [CrossRef]
- Ordyan, M.; Bartol, T.; Kennedy, M.; Rangamani, P.; Sejnowski, T. Interactions between calmodulin and neurogranin govern the dynamics of CaMKII as a leaky integrator. PLoS Comput. Biol. 2020, 16, e1008015. [Google Scholar] [CrossRef]
- Xue, R.; Meng, H.; Yin, J.; Xia, J.; Hu, Z.; Liu, H. The role of calmodulin vs. synaptotagmin in exocytosis. Front. Mol. Neurosci. 2021, 14, 691363. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, J.; Kim, H.Y.; Ryoo, N.; Sejin Lee, S.; Kim, Y.S.; Rhim, H.; Shin, Y.-K. Amyloid-β oligomers may impair SNARE-mediated exocytosis by direct binding to syntaxin 1a. Cell Rep. 2015, 12, 1244–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baracaldo-Santamaría, D.; Avendaño-Lopez, S.S.; Ariza-Salamanca, D.F.; Rodriguez-Giraldo, M.; Calderon-Ospina, C.A.; González-Reyes, R.E.; Nava-Mesa, M.O. Role of calcium modulation in the pathophysiology and treatment of Alzheimer’s disease. Int. J. Mol. Sci. 2023, 24, 9067. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The amyloid-β oligomer hypothesis: Beginning of the third decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [Green Version]
- Morley, J.E.; Farr, S.A.; Nguyen, A.D.; Xu, F. What is the physiological function of amyloid-beta protein? J. Nutr. Health Aging 2019, 23, 225–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Li, L.; Sang, S.; Pan, X.; Zhong, C. Physiological roles of β-amyloid in regulating synaptic function: Implications for AD pathophysiology. Neurosci. Bull. 2022. [Google Scholar] [CrossRef] [PubMed]
- Nassal, D.; Gratz, D.; Hund, T.J. Challenges and Opportunities for Therapeutic Targeting of Calmodulin Kinase II in Heart. Front. Pharmacol. 2020, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Wanga, P.; Lia, W.; Yanga, Y.; Chenga, N.; Zhanga, Y.; Zhanga, N.; Yina, Y.; Tonga, L.; Lib, Z.; Luoa, J. A polypeptide inhibitor of calcineurin blocks the calcineurin-NFAT signalling pathway in vivo and in vitro. J. Enzyme Inhib. Med. Chem. 2022, 37, 202–210. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Day, D.H. Alzheimer’s Disease beyond Calcium Dysregulation: The Complex Interplay between Calmodulin, Calmodulin-Binding Proteins and Amyloid Beta from Disease Onset through Progression. Curr. Issues Mol. Biol. 2023, 45, 6246-6261. https://doi.org/10.3390/cimb45080393
O’Day DH. Alzheimer’s Disease beyond Calcium Dysregulation: The Complex Interplay between Calmodulin, Calmodulin-Binding Proteins and Amyloid Beta from Disease Onset through Progression. Current Issues in Molecular Biology. 2023; 45(8):6246-6261. https://doi.org/10.3390/cimb45080393
Chicago/Turabian StyleO’Day, Danton H. 2023. "Alzheimer’s Disease beyond Calcium Dysregulation: The Complex Interplay between Calmodulin, Calmodulin-Binding Proteins and Amyloid Beta from Disease Onset through Progression" Current Issues in Molecular Biology 45, no. 8: 6246-6261. https://doi.org/10.3390/cimb45080393