
An Actual Insight into the Pathogenic Pathways of Ankylosing Spondylitis

 , , and
, , and
Abstract
1. Introduction
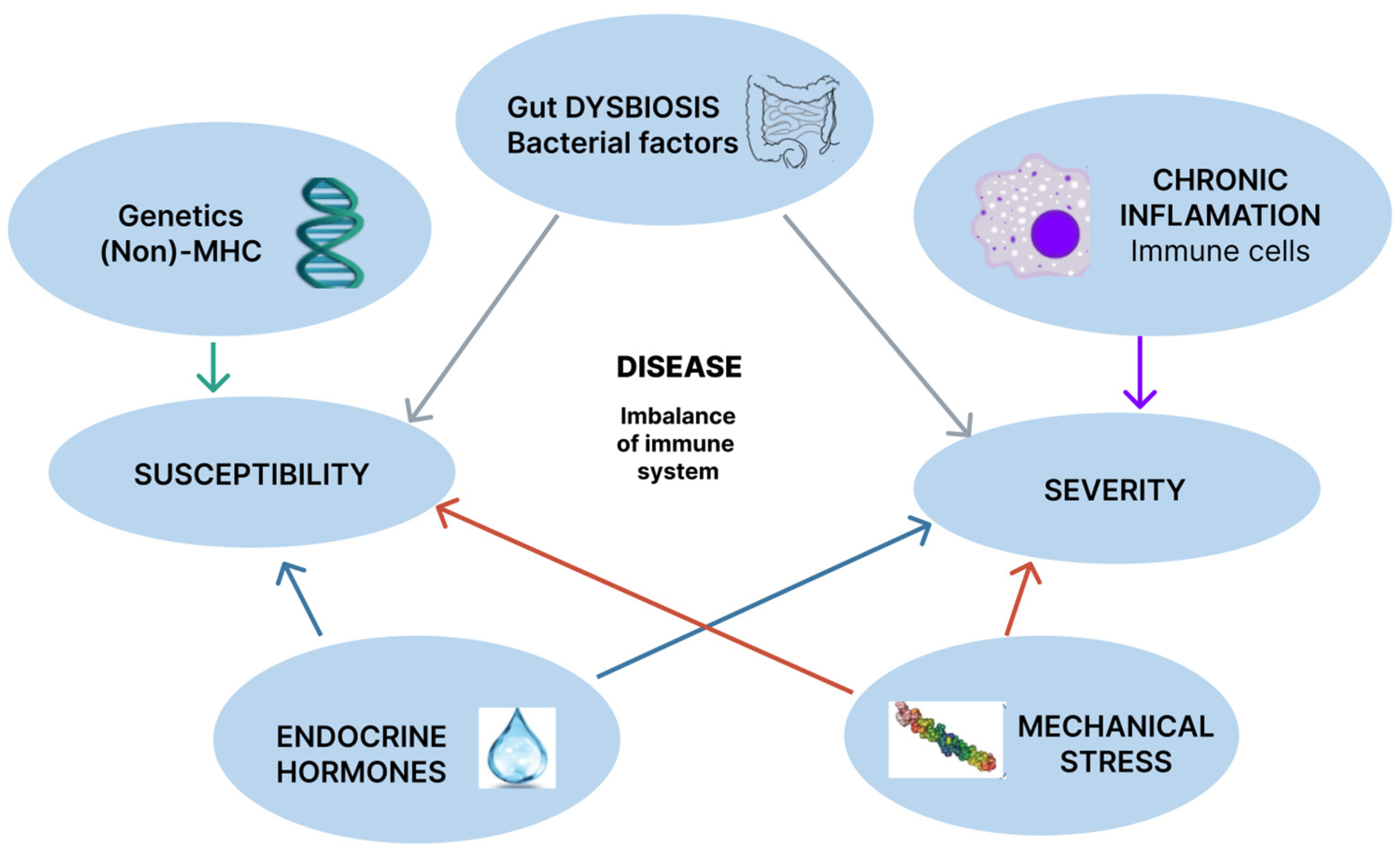
2. Pathogenic Mechanisms
2.1. Genetics
2.1.1. HLA-B27 (MHC Genetics)
2.1.2. MHC Non-HLA-B27
2.1.3. Non-MHC Genetics
ERAP1/ERAP2
Other Non-MHC Genetic Mechanisms
2.2. Environmental Factors
2.2.1. Gut Dysbiosis
2.2.2. Mechanical Stress
2.3. Chronic Inflammation
2.3.1. Immune Cells
2.3.2. The IL-23/IL-17 Axis
2.4. Endocrine Hormones
2.4.1. Vitamin D
2.4.2. Sex Hormones
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Rudwaleit, M.; Landewe, R.; Van Der Heijde, D.; Listing, J.; Brandt, J.; Braun, J.; Chou, C.T.; Collantes-Estevez, E.; Dougados, M.; Huang, F. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part I): Classification of paper patients by expert opinion including uncertainty appraisal. Ann. Rheum. Dis. 2009, 68, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Rudwaleit, M.; Van Der Heijde, D.; Landewe, R.; Listing, J.; Akkoc, N.; Brandt, J.; Braun, J.; Chou, C.T.; Collantes-Estevez, E.; Dougados, M.; et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): Validation and final selection. Ann. Rheum. Dis. 2009, 68, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yuan, Y.; Zhang, S.; Xu, J.; Zou, J. Osteoimmunological insights into the pathogenesis of ankylosing spondylitis. J. Cell. Physiol. 2021, 236, 6090–6100. [Google Scholar] [CrossRef] [PubMed]
- Carli, L.; Calabresi, E.; Governato, G.; Braun, J. One year in review 2018: Axial spondyloarthritis. Clin. Exp. Rheumatol. 2019, 37, 889–898. [Google Scholar]
- Oancea, C.; Mihai, C.; Gherman, D.; Milicescu, M.; Ancuta, I.; Martin, A.; Bojinca, M.; Stoica, V.; Ciuvica, M.M. Development of a prognostic score for work disability in Romanian patients with ankylosing spondylitis. Disabil. Rehabil. 2015, 37, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Coates, L.C. Axial spondyloarthritis and psoriatic arthritis: Mostly overlapping or substantially different diseases? RMD Open 2023, 9, e003063. [Google Scholar] [CrossRef]
- Parameswaran, P.; Lucke, M. HLA-B27 Syndromes; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Hwang, M.C.; Ridley, L.; Reveille, J.D. Ankylosing spondylitis risk factors: A systematic literature review. Clin. Rheumatol. 2021, 40, 3079–3093. [Google Scholar] [CrossRef]
- Kocatürk, B.; Balık, Z.; Pişiren, G.; Kalyoncu, U.; Özmen, F.; Özen, S. Spondyloarthritides: Theories and beyond. Front. Pediatr. 2022, 10, 1074239. [Google Scholar] [CrossRef]
- Bodis, G.; Toth, V.; Schwarting, A. Role of Human Leukocyte Antigens (HLA) in Autoimmune Diseases. Rheumatol. Ther. 2018, 5, 5–20. [Google Scholar] [CrossRef]
- Sharip, A.; Kunz, J. Understanding the Pathogenesis of Spondyloarthritis. Biomolecules 2020, 10, 1461. [Google Scholar] [CrossRef]
- Mathew, A.; Bhagavaldas, M.C.; Biswas, R.; Biswas, L. Genetic risk factors in ankylosing spondylitis: Insights into etiology and dis-ease pathogenesis. Int. J. Rheum. Dis. 2024, 27, e15023. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Xie, P.; Zhou, Z.; Sun, Y.; Wang, F.; You, Y.; Teng, J.; Yang, C.; Zhang, X.; Han, Y. Protective Role of Rheumatic Diseases Against Hepatitis B Virus Infection and Human Leukocyte Antigen B27 Highlighted. Front. Med. 2022, 9, 814423. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernández, A.; Cabrera-Rodríguez, R.; Casado, C.; Pérez-Yanes, S.; Pernas, M.; García-Luis, J.; Marfil, S.; Olivares, I.; Estévez-Herrera, J.; Trujillo-González, R.; et al. Contribution of the HIV-1 Envelope Glycoprotein to AIDS Pathogenesis and Clinical Progression. Biomedicines 2022, 10, 2172. [Google Scholar] [CrossRef]
- Rafiei, A.; Gualandi, M.; Yang, C.-L.; Woods, R.; Kumar, A.; Brunner, K.; Sigrist, J.; Ebersbach, H.; Coats, S.; Renner, C.; et al. IOS-1002, a Stabilized HLA-B57 Open Format, Exerts Potent Anti-Tumor Activity. Cancers 2024, 16, 2902. [Google Scholar] [CrossRef]
- Cortes, A.; Pulit, S.L.; Leo, P.J.; Pointon, J.J.; Robinson, P.C.; Weisman, M.H.; Ward, M.; Gensler, L.S.; Zhou, X.; Garchon, H.J.; et al. Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat. Commun. 2015, 6, 7146. [Google Scholar] [CrossRef]
- Reveille, J.D.; Zhou, X.; Lee, M.; Weisman, M.H.; Yi, L.; Gensler, L.S.; Zou, H.; Ward, M.M.; Ishimori, M.L.; Learch, T.J.; et al. HLA class I and II alleles in susceptibility to ankylosing spondylitis. Ann. Rheum. Dis. 2019, 78, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef]
- Voruganti, A.; Bowness, P. New developments in our understanding of ankylosing spondylitis pathogenesis. Immunology 2020, 161, 94–102. [Google Scholar] [CrossRef]
- Zhu, W.; He, X.; Cheng, K.; Zhang, L.; Chen, D.; Wang, X.; Qiu, G.; Cao, X.; Weng, X. Ankylosing spondylitis: Etiology, pathogenesis, and treatments. Bone Res. 2019, 7, 22. [Google Scholar] [CrossRef]
- Tan, M.; Zhang, Q.; Liu, T.; Yang, Y.; Zheng, J.; Zhou, W.; Xiong, Q.; Qing, Y.F. Autophagy dysfunction may be involved in the pathogenesis of ankylosing spondylitis. Exp. Ther. Med. 2020, 20, 3578–3586. [Google Scholar] [CrossRef] [PubMed]
- Rong, T.; Jia, N.; Wu, B.; Sang, D.; Liu, B. New Insights into the Regulatory Role of Ferroptosis in Ankylosing Spondylitis via Consensus Clustering of Ferroptosis-Related Genes and Weighted Gene Co-Expression Network Analysis. Genes 2022, 13, 1373. [Google Scholar] [CrossRef] [PubMed]
- Dogru, A.; Balkarli, A.; Cetin, G.Y.; Neselioglu, S.; Erel, O.; Tunc, S.E.; Sahin, M. Thiol/disulfide ho-meostasis in patients with ankylosing spondylitis. Bosn. J. Basic Med. Sci. 2016, 16, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Bayır, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef]
- Chang, S.; Tang, M.; Zhang, B.; Xiang, D.; Li, F. Ferroptosis in inflammatory arthritis: A promising future. Front. Immunol. 2022, 13, 955069. [Google Scholar] [CrossRef]
- Li, Q.; Chen, Z.; Yang, C.; Wang, L.; Ma, J.; He, T.; Li, H.; Quan, Z. Role of ferroptosis-associated genes in ankylosing spondylitis and immune cell infiltration. Front. Genet. 2022, 13, 948290. [Google Scholar] [CrossRef]
- McKenzie, B.A.; Dixit, V.M.; Power, C. Fiery Cell Death: Pyroptosis in the Central Nervous System. Trends Neurosci. 2020, 43, 55–73. [Google Scholar] [CrossRef]
- Yang, G.; Kang, H.C.; Cho, Y.-Y.; Lee, H.S.; Lee, J.Y. Inflammasomes and their roles in arthritic disease pathogenesis. Front. Mol. Biosci. 2022, 9, 1027917. [Google Scholar] [CrossRef]
- Yi, Y.-S. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J. Physiol. Pharmacol. 2018, 22, 1. [Google Scholar] [CrossRef]
- Kim, S.-K.; Cho, Y.J.; Choe, J.-Y. NLRP3 inflammasomes and NLRP3 inflammasome-derived proinflammatory cytokines in peripheral blood mononuclear cells of patients with an-kylosing spondylitis. Clin. Chim. Acta 2018, 486, 269–274. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Wang, H.; Zhao, X. Exploring hub pyroptosis-related genes, molecular subtypes, and potential drugs in ankylosing spondylitis by comprehensive bioinformatics analysis and molecular docking. BMC Musculoskelet. Disord. 2023, 24, 532. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.-Y.; Yuan, D.; Zhang, S.-X. Role of the microbiome and its metabolites in ankylosing spondylitis. Front. Immunol. 2022, 13, 1010572. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.-S. Roles of the Caspase-11 Non-Canonical Inflammasome in Rheumatic Diseases. Int. J. Mol. Sci. 2024, 25, 2091. [Google Scholar] [CrossRef]
- Berland, M.; Meslier, V.; Berreira Ibraim, S.; Le Chatelier, E.; Pons, N.; Maziers, N.; Thirion, F.; Gauthier, F.; Plaza Oñate, F.; Furet, J.P.; et al. Both Disease Activity and HLA–B27 Status Are Associated With Gut Microbiome Dysbiosis in Spondyloarthritis Patients. Arthritis Rheumatol. 2023, 75, 41–52. [Google Scholar] [CrossRef]
- Xiong, Y.; Cai, M.; Xu, Y.; Dong, P.; Chen, H.; He, W.; Zhang, J. Joint together: The etiology and pathogenesis of ankylosing spondylitis. Front. Immunol. 2022, 13, 996103. [Google Scholar] [CrossRef]
- Antoniou, A.N.; Lenart, I.; Kriston-Vizi, J.; Iwawaki, T.; Turmaine, M.; McHugh, K.; Ali, S.; Blake, N.; Bowness, P.; Bajaj-Elliott, M.; et al. Salmonella exploits HLA-B27 and host unfolded protein responses to promote intracellular replication. Ann. Rheum. Dis. 2019, 78, 74–82. [Google Scholar] [CrossRef]
- Breban, M.; Tap, J.; Leboime, A.; Said-Nahal, R.; Langella, P.; Chiocchia, G.; Furet, J.P.; Sokol, H. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann. Rheum. Dis. 2017, 76, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.C.-C.; Chou, M.-C.; Huang, J.-Y.; Chang, R.; Hung, Y.-M. The association between Candida infection and ankylosing spondylitis: A population-based matched cohort study. Curr. Med. Res. Opin. 2020, 36, 2063–2069. [Google Scholar] [CrossRef]
- Mauro, D.; Simone, D.; Bucci, L.; Ciccia, F. Novel immune cell phenotypes in spondyloarthritis pathogenesis. Semin. Immunopathol. 2021, 43, 265–277. [Google Scholar] [CrossRef]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Saieva, L.; Peralta, S.; Giardina, A.; Cannizzaro, A.; Sireci, G.; De Leo, G.; Alessandro, R.; et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann. Rheum. Dis. 2015, 74, 1739–1747. [Google Scholar] [CrossRef]
- Ciccia, F.; Bombardieri, M.; Principato, A.; Giardina, A.; Tripodo, C.; Porcasi, R.; Peralta, S.; Franco, V.; Giardina, E.; Craxi, A.; et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 2009, 60, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Wordsworth, B.P.; Cohen, C.J.; Davidson, C.; Vecellio, M. Perspectives on the Genetic Associations of Ankylosing Spondylitis. Front. Immunol. 2021, 12, 603726. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.-C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4−CD8− entheseal resident T cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef]
- Cuthbert, R.J.; Watad, A.; Fragkakis, E.M.; Dunsmuir, R.; Loughenbury, P.; Khan, A.; Millner, P.A.; Davison, A.; Marzo-Ortega, H.; Newton, D.; et al. Evidence that tissue resident human enthesis γδT-cells can produce IL-17A independently of IL-23R transcript expression. Ann. Rheum. Dis. 2019, 78, 1559–1565. [Google Scholar] [CrossRef]
- Kim, T.; Stone, M.; Payne, U.; Zhang, X.; Ionescu, M.; Lobanok, T.; King, L.; Poole, A.R.; Inman, R.D. Cartilage biomarkers in ankylosing spondylitis: Relationship to clinical variables and treatment response. Arthritis Rheum. 2005, 52, 885–891. [Google Scholar] [CrossRef]
- Rezaiemanesh, A.; Abdolmaleki, M.; Abdolmohammadi, K.; Aghaei, H.; Pakdel, F.D.; Fatahi, Y.; Soleimanifar, N.; Zavvar, M.; Nicknam, M.H. Immune cells involved in the pathogenesis of ankylosing spondylitis. Biomed. Pharmacother. 2018, 100, 198–204. [Google Scholar] [CrossRef]
- Martínez-Ramos, S.; Rafael-Vidal, C.; Pego-Reigosa, J.M.; García, S. Monocytes and Macrophages in Spondyloarthritis: Functional Roles and Effects of Current Therapies. Cells 2022, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, E.; Baeten, D.; De Rycke, L.; Vandooren, B.; Foell, D.; Roth, J.; Cañete, J.D.; Boots, A.M.; Veys, E.M.; De Keyser, F. Synovial histopathology of psoriatic arthritis, both oligo- and polyarticular, resembles spondyloarthropathy more than it does rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R569. [Google Scholar] [CrossRef]
- Braun Jür Bollow, M.; Neure, L.; Seipelt, E.; Seyrekbasan, F.; Herbst, H.; Eggens, U.; Distler, A.; Sieper, J. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum. 1995, 38, 499–505. [Google Scholar] [CrossRef]
- Azuz-Lieberman, N.; Markel, G.; Mizrahi, S.; Gazit, R.; Hanna, J.; Achdout, H.; Gruda, R.; Katz, G.; Arnon, T.I.; Battat, S.; et al. The involvement of NK cells in ankylosing spondylitis. Int. Immunol. 2005, 17, 837–845. [Google Scholar] [CrossRef]
- Kucuksezer, U.C.; Aktas Cetin, E.; Esen, F.; Tahrali, I.; Akdeniz, N.; Gelmez, M.Y.; Deniz, G. The Role of Natural Killer Cells in Autoimmune Diseases. Front. Immunol. 2021, 12, 622306. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Van Den Bosch, F. IL-23 and axial disease: Do they come together? Rheumatology 2021, 60, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Tsukazaki, H.; Kaito, T. The Role of the IL-23/IL-17 Pathway in the Pathogenesis of Spondyloarthritis. Int. J. Mol. Sci. 2020, 21, 6401. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, Q.; Wang, G.; Yang, C.; Xu, Y.; Li, Y.; Yang, P. Circulating levels of Th1 and Th2 chemokines in patients with ankylosing spondylitis. Cytokine 2016, 81, 10–14. [Google Scholar] [CrossRef]
- Rotondi, M.; Rosati, A.; Buonamano, A.; Lasagni, L.; Lazzeri, E.; Pradella, F.; Fossombroni, V.; Cirami, C.; Liotta, F.; La Villa, G.; et al. High Pretransplant Serum Levels of CXCL10/IP-10 Are Related to Increased Risk of Renal Allograft Failure. Am. J. Transplant. 2004, 4, 1466–1474. [Google Scholar] [CrossRef]
- Schinocca, C.; Rizzo, C.; Fasano, S.; Grasso, G.; La Barbera, L.; Ciccia, F.; Guggino, G. Role of the IL-23/IL-17 Pathway in Rheumatic Diseases: An Overview. Front. Immunol. 2021, 12, 637829. [Google Scholar] [CrossRef]
- Cutolo, M.; Otsa, K.; Paolino, S.; Yprus, M.; Veldi, T.; Seriolo, B. Vitamin D involvement in rheumatoid arthritis and systemic lupus erythaematosus. Ann. Rheum. Dis. 2009, 68, 446–447. [Google Scholar] [CrossRef]
- Cai, G.; Wang, L.; Fan, D.; Xin, L.; Liu, L.; Hu, Y.; Ding, N.; Xu, S.; Xia, G.; Jin, X.; et al. Vitamin D in ankylosing spondylitis: Review and meta-analysis. Clin. Chim. Acta 2015, 438, 316–322. [Google Scholar] [CrossRef]
- Shany, S.; Levy, Y.; Lahav-Cohen, M. The effects of 1α,24(S)-dihydroxyvitamin D2 analog on cancer cell proliferation and cytokine expression. Steroids 2001, 66, 319–325. [Google Scholar] [CrossRef]
- Popescu, M.; Feldman, T.B.; Chitnis, T. Interplay Between Endocrine Disruptors and Immunity: Implications for Diseases of Autoreactive Etiology. Front. Pharmacol. 2021, 12, 626107. [Google Scholar] [CrossRef]
- Aurelian, S.; Ciobanu, A.; Cărare, R.; Stoica, S.-I.; Anghelescu, A.; Ciobanu, V.; Onose, G.; Munteanu, C.; Popescu, C.; Andone, I.; et al. Topical Cellular/Tissue and Molecular Aspects Regarding Nonpharmacological Inter-ventions in Alzheimer’s Disease—A Systematic Review. Int. J. Mol. Sci. 2023, 24, 16533. [Google Scholar] [CrossRef] [PubMed]
- Aydin, T.; Karacan, İ.; Demir, S.E.; Sahin, Z. Bone loss in males with ankylosing spondylitis: Its relation to sex hormone levels. Clin. Endocrinol. 2005, 63, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, E.; Adami, G.; Rossini, M.; Giollo, A.; Orsolini, G.; Viapiana, O.; Gatti, D.; Fassio, A. The Emerging Roles of Endocrine Hormones in Different Arthritic Disorders. Front. Endocrinol. 2021, 12, 620920. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Type of Mechanism | Marker | Basis | Pathway | Role |
|---|---|---|---|---|
| Genetics | HLA-B27 and other MHC alleles | Polymorphism | Peptide binding specificity, misfolding, and forming heavy chain homodimers; | Susceptibility |
| Non-MHC—ERAP1 | Polymorphism | Alterations in peptide and cytokine receptors processing | Susceptibility | |
| Gut dysbiosis | Ruminococcus gnavus and species of Clostridiales (Erysipelatoclostridium ramosum, Clostridium symbiosum, Clostridium bolteae) | PAMPs, DAMPs | Changes in the gut microbiome, inappropriate immune responses | Susceptibility and severity |
| Mechanical stress | CD3+CD4-CD8-type expressing IL-23R lymphocytes, Epitopes from type II collagen degradation | Microtraumas releasing arthritogenic peptides | Dysfunction in the TNF-α or IL-23/IL-17 pathway | Trigger and disease activity |
| Chronic inflammation | High Th1 density, Increased Th1/Th2 fraction, IL-23/IL-17 pathway, IL-23R genetic polymorphism | Changes in cell response | Activation and accumulation of immune cells (dendritic cells, macrophages, and natural killer cells) with the secretion of cytokines | Progression and severity |
| Endocrine hormones | Sex hormones, Vitamin D, Modulators of the Wnt pathway | Effects on the immune system | Regulating the balance of Th1/Th2 and TNF-α production | Onset, disease activity, and severity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Păsăran, E.-D.; Diaconu, A.E.; Oancea, C.; Bălănescu, A.-R.; Aurelian, S.M.; Homentcovschi, C. An Actual Insight into the Pathogenic Pathways of Ankylosing Spondylitis. Curr. Issues Mol. Biol. 2024, 46, 12800-12812. https://doi.org/10.3390/cimb46110762
Păsăran E-D, Diaconu AE, Oancea C, Bălănescu A-R, Aurelian SM, Homentcovschi C. An Actual Insight into the Pathogenic Pathways of Ankylosing Spondylitis. Current Issues in Molecular Biology. 2024; 46(11):12800-12812. https://doi.org/10.3390/cimb46110762
Chicago/Turabian StylePăsăran, Emilia-Daniela, Andreea Elena Diaconu, Corina Oancea, Andra-Rodica Bălănescu, Sorina Maria Aurelian, and Corina Homentcovschi. 2024. "An Actual Insight into the Pathogenic Pathways of Ankylosing Spondylitis" Current Issues in Molecular Biology 46, no. 11: 12800-12812. https://doi.org/10.3390/cimb46110762
APA StylePăsăran, E.-D., Diaconu, A. E., Oancea, C., Bălănescu, A.-R., Aurelian, S. M., & Homentcovschi, C. (2024). An Actual Insight into the Pathogenic Pathways of Ankylosing Spondylitis. Current Issues in Molecular Biology, 46(11), 12800-12812. https://doi.org/10.3390/cimb46110762