
The Vitamin K-Dependent Anticoagulant Factor, Protein S, Regulates Vascular Permeability

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Materials
2.3. Transwell Permeability Assay
2.4. VEC Internalization Assay
2.5. VEC Cleavage Assay
2.6. Western Blotting Assay
2.7. Actin and VEC Immunostaining
2.8. Determination of Length of VEC Membrane Stretches
2.9. Determination of the Intracellular Fraction of VEC
2.10. Statistical Analysis
3. Results
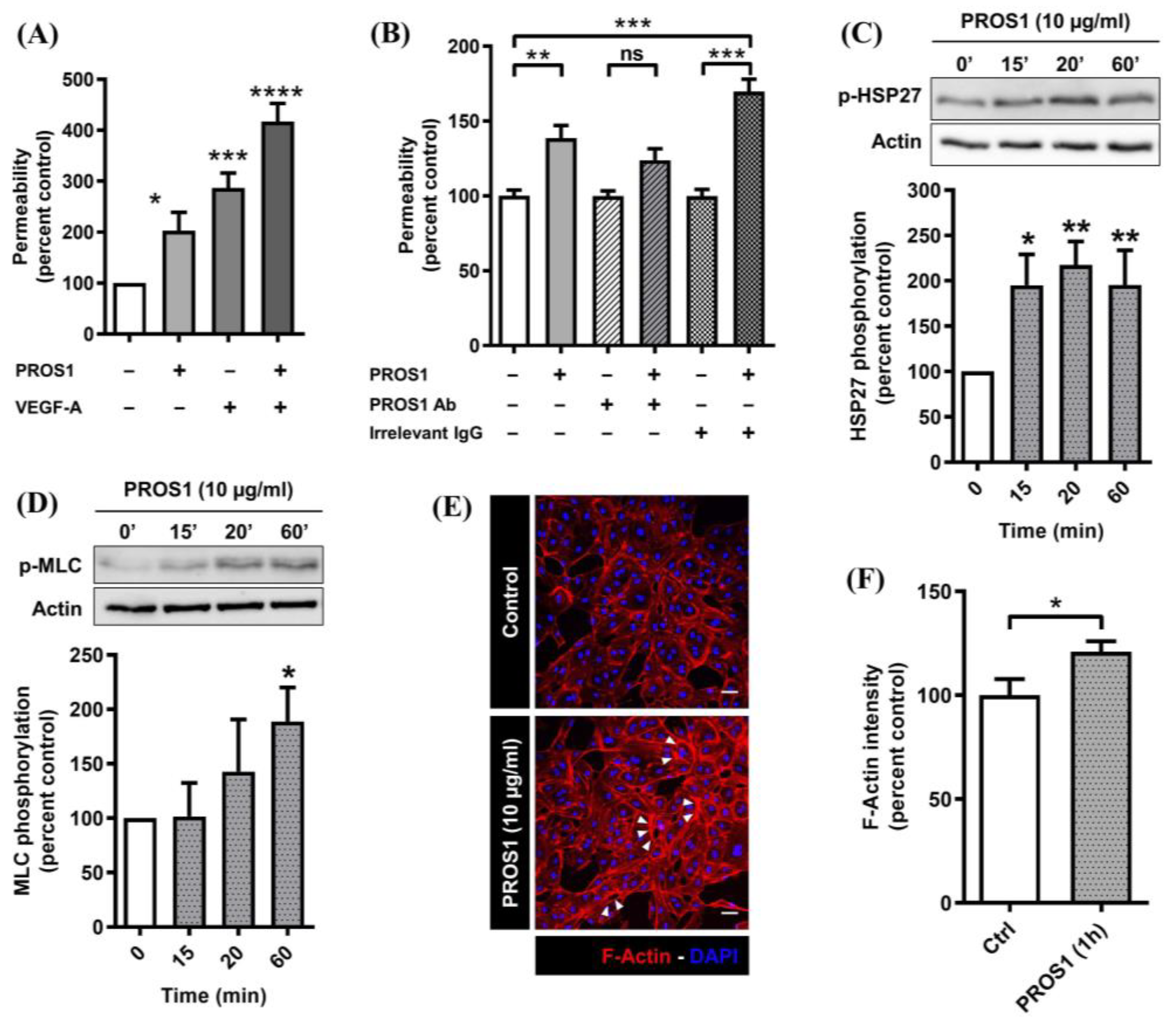
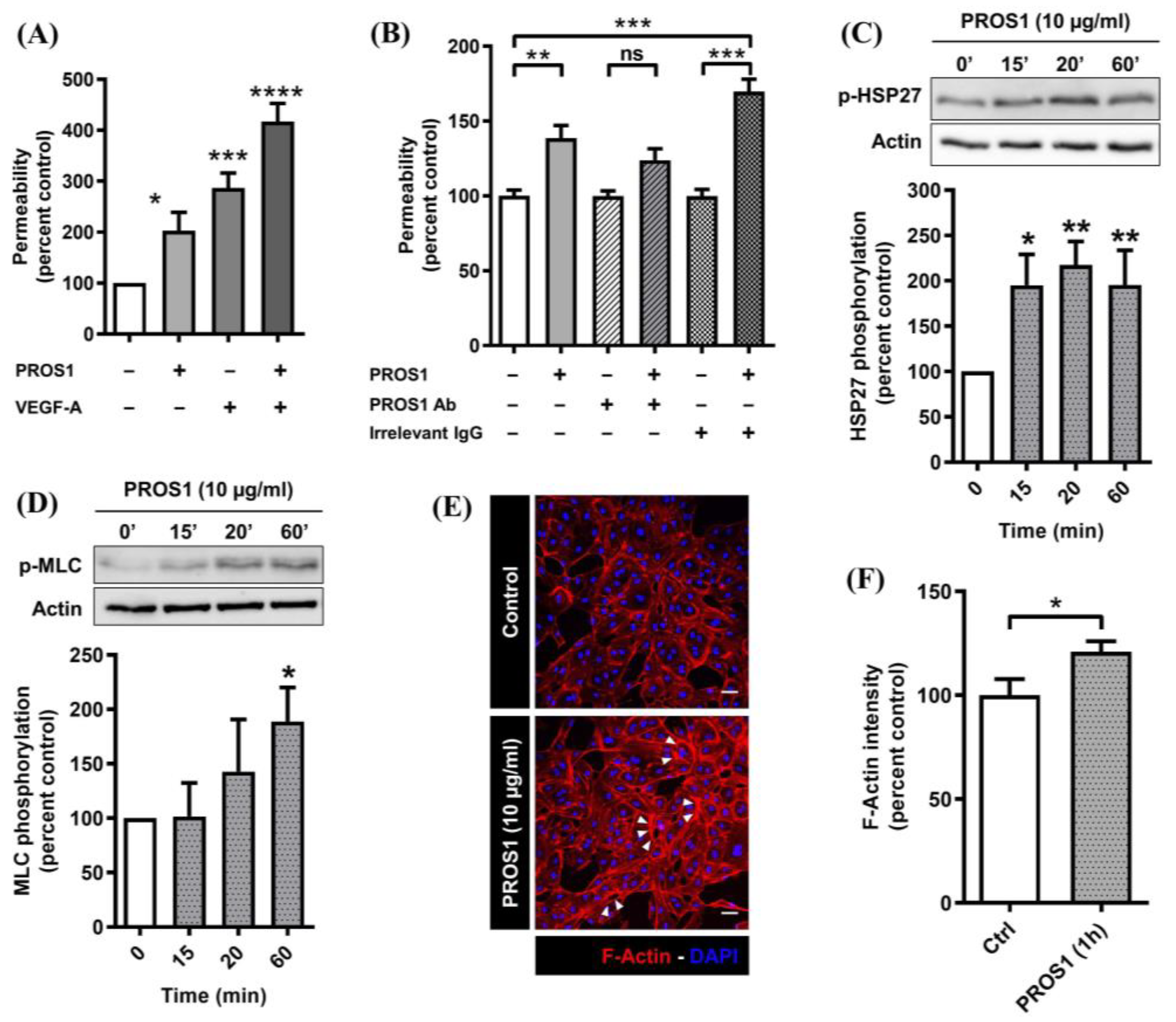
3.1. Human PROS1 Increases Endothelial Cell Monolayer Permeability and Activates the p38 MAPK and Rho/ROCK Pathways
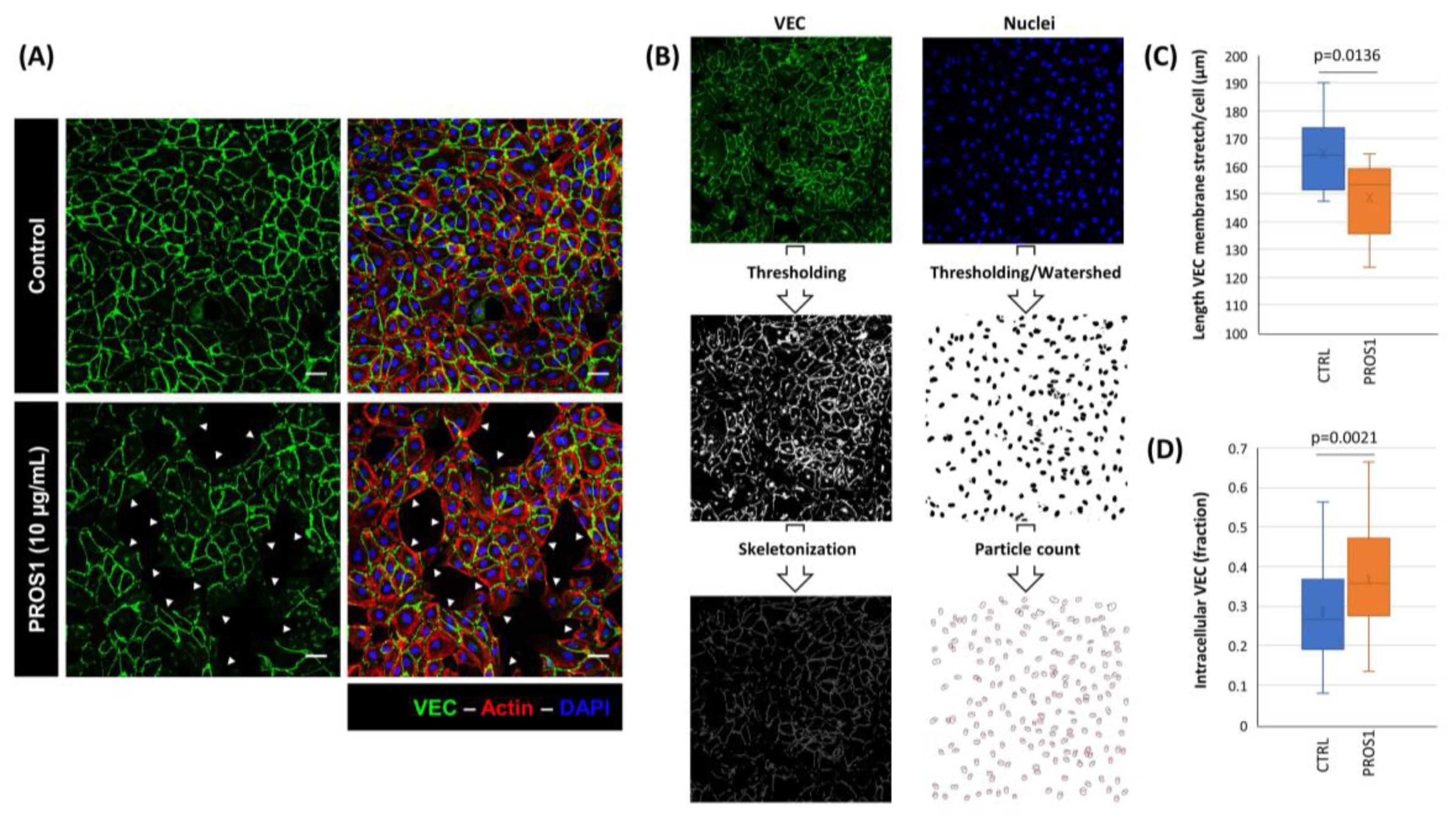
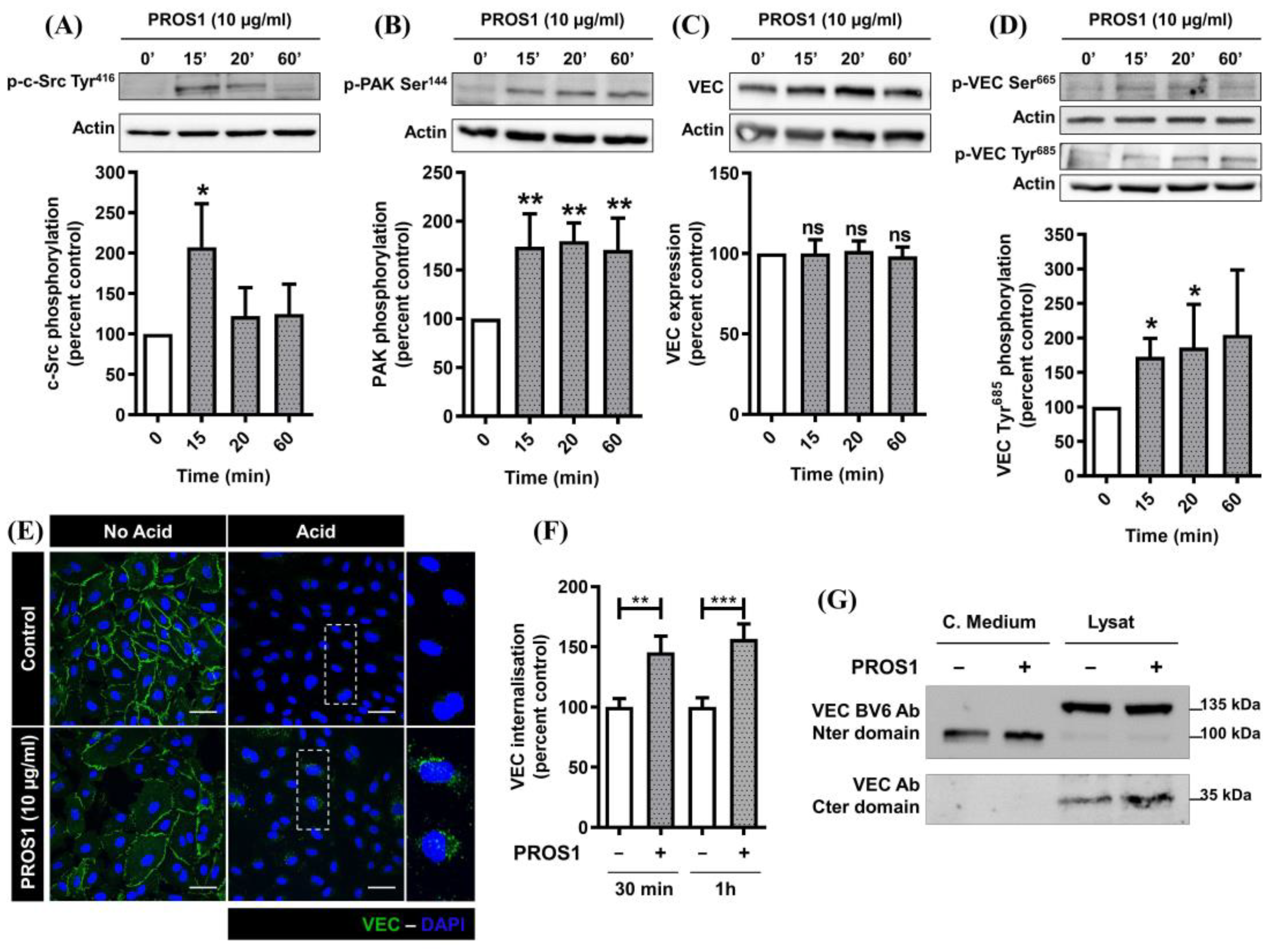
3.2. Human PROS1 Regulates Vascular Endothelial Cadherin (VEC) Internalization and Cleavage
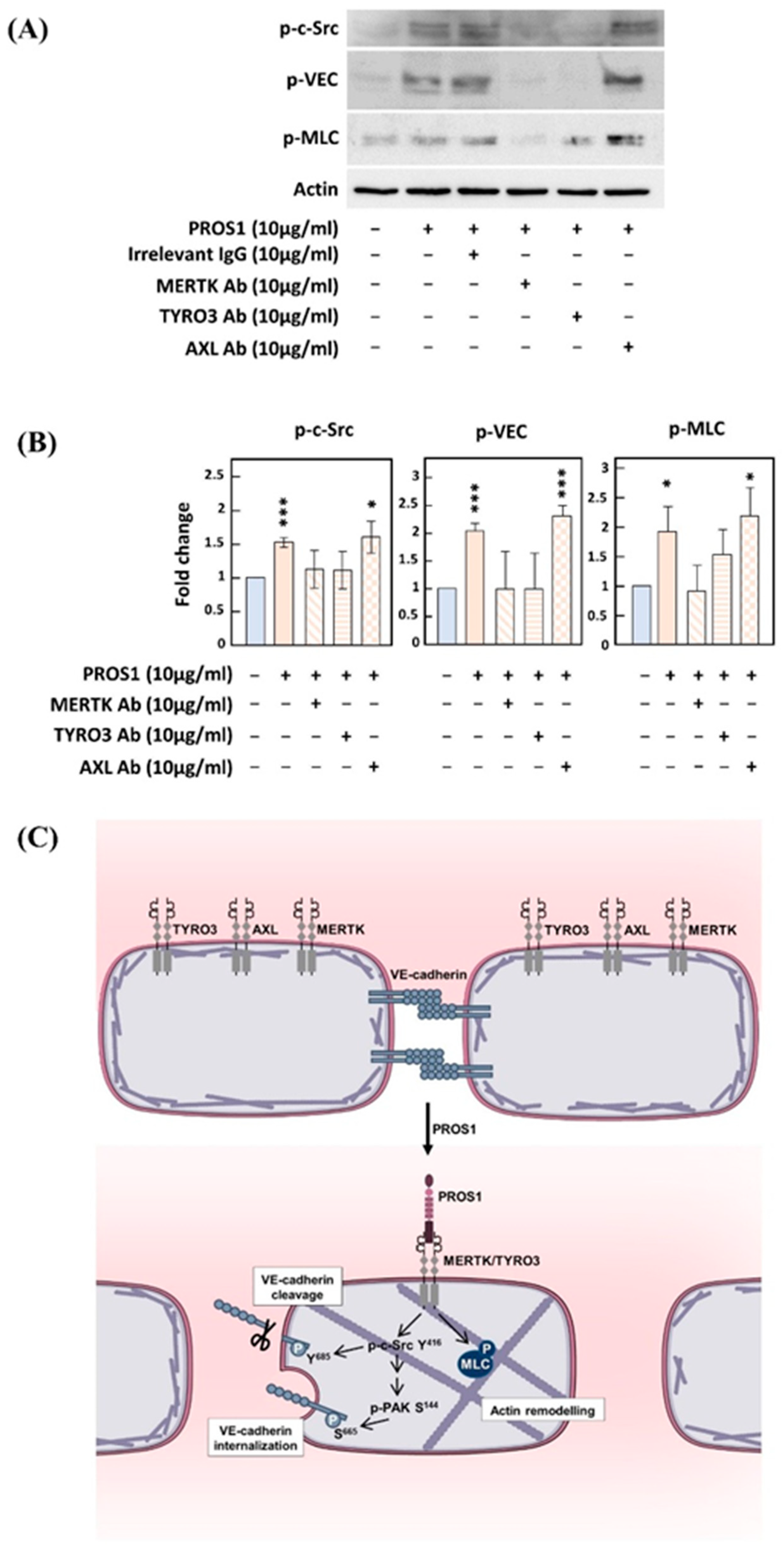
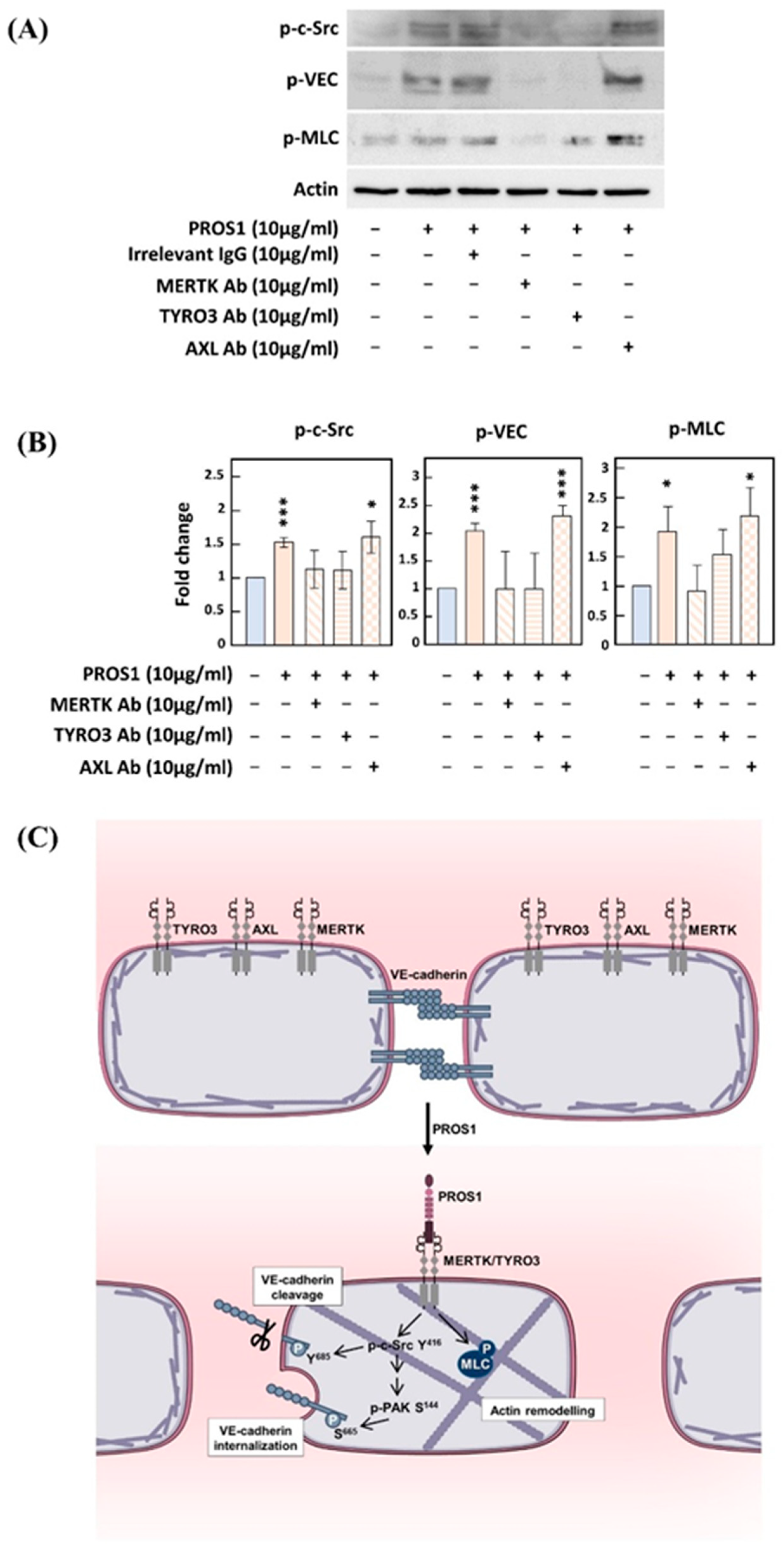
3.3. Implication of Both MERTK and TYRO3 Receptors in PROS1-Induced c-Src and VEC Phosphorylation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saller, F. The-Carboxyglutamic Acid Domain of Anticoagulant Protein S Is Involved in Activated Protein C Cofactor Activity, Independently of Phospholipid Binding. Blood 2005, 105, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Dahlbäck, B. Vitamin K–Dependent Protein S: Beyond the Protein C Pathway. Semin. Thromb. Hemost. 2018, 44, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Dahlbäck, B. The Tale of Protein S and C4b-Binding Protein, a Story of Affection. Thromb. Haemost. 2007, 98, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Lumbroso, D.; Soboh, S.; Maimon, A.; Schif-Zuck, S.; Ariel, A.; Burstyn-Cohen, T. Macrophage-Derived Protein S Facilitates Apoptotic Polymorphonuclear Cell Clearance by Resolution Phase Macrophages and Supports Their Reprogramming. Front. Immunol. 2018, 9, 358. [Google Scholar] [CrossRef] [PubMed]
- Fair, D.S.; Marlar, R.A.; Levin, E.G. Human Endothelial Cells Synthesize Protein S. Blood 1986, 67, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Benzakour, O.; Kanthou, C. The Anticoagulant Factor, Protein S, Is Produced by Cultured Human Vascular Smooth Muscle Cells and Its Expression Is up-Regulated by Thrombin. Blood 2000, 95, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Tsou, W.-I.; Nguyen, K.-Q.N.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor Tyrosine Kinases, TYRO3, AXL, and MER, Demonstrate Distinct Patterns and Complex Regulation of Ligand-Induced Activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, Y.; Singh, I.; Bell, R.D.; Deane, R.; Zhong, Z.; Sagare, A.; Winkler, E.A.; Zlokovic, B.V. Protein S Controls Hypoxic/Ischemic Blood-Brain Barrier Disruption through the TAM Receptor Tyro3 and Sphingosine 1-Phosphate Receptor. Blood 2010, 115, 4963–4972. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Barrett, T.M.; Zhong, Z.; Fernández, J.A.; Griffin, J.H.; Freeman, R.S.; Zlokovic, B.V. Protein S Blocks the Extrinsic Apoptotic Cascade in Tissue Plasminogen Activator/N-Methyl D-Aspartate-Treated Neurons via Tyro3-Akt-FKHRL1 Signaling Pathway. Mol. Neurodegener. 2011, 6, 13. [Google Scholar] [CrossRef]
- Liao, D.; Wang, X.; Li, M.; Lin, P.H.; Yao, Q.; Chen, C. Human Protein S Inhibits the Uptake of AcLDL and Expression of SR-A through Mer Receptor Tyrosine Kinase in Human Macrophages. Blood 2009, 113, 165–174. [Google Scholar] [CrossRef]
- Fraineau, S.; Monvoisin, A.; Clarhaut, J.; Talbot, J.; Simonneau, C.; Kanthou, C.; Kanse, S.M.; Philippe, M.; Benzakour, O. The Vitamin K–Dependent Anticoagulant Factor, Protein S, Inhibits Multiple VEGF-A–Induced Angiogenesis Events in a Mer-and SHP2-Dependent Manner. Blood 2012, 120, 5073–5083. [Google Scholar] [CrossRef]
- Prasad, D.; Rothlin, C.V.; Burrola, P.; Burstyn-Cohen, T.; Lu, Q.; Garcia de Frutos, P.; Lemke, G. TAM Receptor Function in the Retinal Pigment Epithelium. Mol. Cell. Neurosci. 2006, 33, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.A.; Maylock, C.A.; Williams, J.A.; Paweletz, C.P.; Shu, H.; Shacter, E. Serum-Derived Protein S Binds to Phosphatidylserine and Stimulates the Phagocytosis of Apoptotic Cells. Nat. Immunol. 2003, 4, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Gely-Pernot, A.; Coronas, V.; Harnois, T.; Prestoz, L.; Mandairon, N.; Didier, A.; Berjeaud, J.M.; Monvoisin, A.; Bourmeyster, N.; De Frutos, P.G.; et al. An Endogenous Vitamin K-Dependent Mechanism Regulates Cell Proliferation in the Brain Subventricular Stem Cell Niche. Stem Cells 2012, 30, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Yefimova, M.G.; Messaddeq, N.; Harnois, T.; Meunier, A.-C.; Clarhaut, J.; Noblanc, A.; Weickert, J.-L.; Cantereau, A.; Philippe, M.; Bourmeyster, N.; et al. A Chimerical Phagocytosis Model Reveals the Recruitment by Sertoli Cells of Autophagy for the Degradation of Ingested Illegitimate Substrates. Autophagy 2013, 9, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Ginisty, A.; Gély-Pernot, A.; Abaamrane, L.; Morel, F.; Arnault, P.; Coronas, V.; Benzakour, O. Evidence for a Subventricular Zone Neural Stem Cell Phagocytic Activity Stimulated by the Vitamin K-Dependent Factor Protein S: Phagocytic Activity of Neural Stem Cells. Stem Cells 2015, 33, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Abboud-Jarrous, G.; Priya, S.; Maimon, A.; Fischman, S.; Cohen-Elisha, M.; Czerninski, R.; Burstyn-Cohen, T. Protein S Drives Oral Squamous Cell Carcinoma Tumorigenicity through Regulation of AXL. Oncotarget 2017, 8, 13986. [Google Scholar] [CrossRef]
- Zelentsova, K.; Talmi, Z.; Abboud-Jarrous, G.; Sapir, T.; Capucha, T.; Nassar, M.; Burstyn-Cohen, T. Protein S Regulates Neural Stem Cell Quiescence and Neurogenesis: Protein S in NSC Quiescence and Neurogenesis. Stem Cells 2017, 35, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Burstyn-Cohen, T.; Fresia, R. TAM Receptors in Phagocytosis: Beyond the Mere Internalization of Particles. Immunol. Rev. 2023, 319, 7–26. [Google Scholar] [CrossRef]
- Collett, G.; Wood, A.; Alexander, M.Y.; Varnum, B.C.; Boot-Handford, R.P.; Ohanian, V.; Ohanian, J.; Fridell, Y.-W.; Canfield, A.E. Receptor Tyrosine Kinase Axl Modulates the Osteogenic Differentiation of Pericytes. Circ. Res. 2003, 92, 1123–1129. [Google Scholar] [CrossRef]
- McShane, L.; Tabas, I.; Lemke, G.; Kurowska-Stolarska, M.; Maffia, P. TAM Receptors in Cardiovascular Disease. Cardiovasc. Res. 2019, 115, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Burstyn-Cohen, T.; Heeb, M.J.; Lemke, G. Lack of Protein S in Mice Causes Embryonic Lethal Coagulopathy and Vascular Dysgenesis. J. Clin. Investig. 2009, 119, 2942–2953. [Google Scholar] [CrossRef] [PubMed]
- Saller, F.; Brisset, A.C.; Tchaikovski, S.N.; Azevedo, M.; Chrast, R.; Fernandez, J.A.; Schapira, M.; Hackeng, T.M.; Griffin, J.H.; Angelillo-Scherrer, A. Generation and Phenotypic Analysis of Protein S-Deficient Mice. Blood 2009, 114, 2307–2314. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, K.; Molina, A.J.A.; Guo, W.; Roy, S. High Glucose Disrupts Mitochondrial Morphology in Retinal Endothelial Cells: Implications for Diabetic Retinopathy. Am. J. Pathol. 2010, 177, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhan, H.; Zhou, X.-Y.; Yao, L.; Yan, M.; Chen, A.; Liu, J.; Ren, X.; Zhang, X.; Liu, J.-X.; et al. MicroRNA-22 Regulates Inflammation and Angiogenesis via Targeting VE-Cadherin. FEBS Lett. 2017, 591, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling Mechanisms Regulating Endothelial Permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- García-Ponce, A.; Citalán-Madrid, A.F.; Velázquez-Avila, M.; Vargas-Robles, H.; Schnoor, M. The Role of Actin-Binding Proteins in the Control of Endothelial Barrier Integrity. Thromb. Haemost. 2015, 113, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Adamson, R.H.; Curry, F.E.; Adamson, G.; Liu, B.; Jiang, Y.; Aktories, K.; Barth, H.; Daigeler, A.; Golenhofen, N.; Ness, W.; et al. Rho and Rho Kinase Modulation of Barrier Properties: Cultured Endothelial Cells and Intact Microvessels of Rats and Mice. J. Physiol. 2002, 539, 295–308. [Google Scholar] [CrossRef]
- Goldberg, P.L.; MacNaughton, D.E.; Clements, R.T.; Minnear, F.L.; Vincent, P.A. P38 MAPK Activation by TGF-Β1 Increases MLC Phosphorylation and Endothelial Monolayer Permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L146–L154. [Google Scholar] [CrossRef]
- Desai, R.; Sarpal, R.; Ishiyama, N.; Pellikka, M.; Ikura, M.; Tepass, U. Monomeric α-Catenin Links Cadherin to the Actin Cytoskeleton. Nat. Cell Biol. 2013, 15, 261–273. [Google Scholar] [CrossRef]
- Navaratna, D.; McGuire, P.G.; Menicucci, G.; Das, A. Proteolytic Degradation of VE-Cadherin Alters the Blood-Retinal Barrier in Diabetes. Diabetes 2007, 56, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. ADAM10 Regulates Endothelial Permeability and T-Cell Transmigration by Proteolysis of Vascular Endothelial Cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, C.; Klitgaard, M.; Noer, J.B.; Kotzsch, A.; Nehammer, C.; Kronqvist, P.; Berthelsen, J.; Blobel, C.; Kveiborg, M.; Albrechtsen, R.; et al. ADAM12 Is Expressed in the Tumour Vasculature and Mediates Ectodomain Shedding of Several Membrane-Anchored Endothelial Proteins. Biochem. J. 2013, 452, 97–109. [Google Scholar] [CrossRef]
- Chong, C.; Tan, L.; Lim, L.; Manser, E. The Mechanism of PAK Activation. Autophosphorylation Events in Both Regulatory and Kinase Domains Control Activity. J. Biol. Chem. 2001, 276, 17347–17353. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J.; Gutkind, J.S. VEGF Controls Endothelial-Cell Permeability by Promoting the β-Arrestin-Dependent Endocytosis of VE-Cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Rane, C.K.; Minden, A. P21 Activated Kinases. Small GTPases 2014, 5, e28003. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Sanawar, R.; Li, X.; Li, F. Structure, Biochemistry, and Biology of PAK Kinases. Gene 2017, 605, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Cand, F.; Cruzalegui, F.; Wernstedt, C.; Souchelnytskyi, S.; Vilgrain, I.; Huber, P. Src Kinase Phosphorylates Vascular Endothelial-Cadherin in Response to Vascular Endothelial Growth Factor: Identification of Tyrosine 685 as the Unique Target Site. Oncogene 2007, 26, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Sidibé, A.; Mannic, T.; Arboleas, M.; Subileau, M.; Gulino-Debrac, D.; Bouillet, L.; Jan, M.; Vandhuick, T.; Le Loët, X.; Vittecoq, O.; et al. Soluble VE-Cadherin in Rheumatoid Arthritis Patients Correlates with Disease Activity: Evidence for Tumor Necrosis Factor α-Induced VE-Cadherin Cleavage. Arthritis Rheum. 2012, 64, 77–87. [Google Scholar] [CrossRef]
- Sidibé, A.; Polena, H.; Pernet-Gallay, K.; Razanajatovo, J.; Mannic, T.; Chaumontel, N.; Bama, S.; Maréchal, I.; Huber, P.; Gulino-Debrac, D.; et al. VE-Cadherin Y685F Knock-in Mouse Is Sensitive to Vascular Permeability in Recurrent Angiogenic Organs. Am. J. Physiol.-Heart Circ. Physiol. 2014, 307, H455–H463. [Google Scholar] [CrossRef]
- Kanthou, C. The Tumor Vascular Targeting Agent Combretastatin A-4-Phosphate Induces Reorganization of the Actin Cytoskeleton and Early Membrane Blebbing in Human Endothelial Cells. Blood 2002, 99, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.W.; Holmes, T.; Fisher, M.; Tozer, G.M.; Harrity, J.P.A.; Kanthou, C. Evaluation of Sydnone-Based Analogues of Combretastatin A-4 Phosphate (CA4P) as Vascular Disrupting Agents for Use in Cancer Therapy. ChemMedChem 2018, 13, 2618–2626. [Google Scholar] [CrossRef] [PubMed]
- Polena, H.; Creuzet, J.; Dufies, M.; Sidibé, A.; Khalil-Mgharbel, A.; Salomon, A.; Deroux, A.; Quesada, J.-L.; Roelants, C.; Filhol, O.; et al. The Tyrosine-Kinase Inhibitor Sunitinib Targets Vascular Endothelial (VE)-Cadherin: A Marker of Response to Antitumoural Treatment in Metastatic Renal Cell Carcinoma. Br. J. Cancer 2018, 118, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Dahlback, B. Blood Coagulation and Its Regulation by Anticoagulant Pathways: Genetic Pathogenesis of Bleeding and Thrombotic Diseases. J. Intern. Med. 2005, 257, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.A.; Key, N.S.; Levy, J.H. Blood Coagulation: Hemostasis and Thrombin Regulation. Anesth. Analg. 2009, 108, 1433–1446. [Google Scholar] [CrossRef] [PubMed]
- van Hinsbergh, V.W.M. Endothelium—Role in Regulation of Coagulation and Inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial Cell Control of Thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [PubMed]
- van Nieuw Amerongen, G.P.; Musters, R.J.P.; Eringa, E.C.; Sipkema, P.; van Hinsbergh, V.W.M. Thrombin-Induced Endothelial Barrier Disruption in Intact Microvessels: Role of RhoA/Rho Kinase-Myosin Phosphatase Axis. Am. J. Physiol. Cell Physiol. 2008, 294, C1234–C1241. [Google Scholar] [CrossRef] [PubMed]
- Mikelis, C.M.; Simaan, M.; Ando, K.; Fukuhara, S.; Sakurai, A.; Amornphimoltham, P.; Masedunskas, A.; Weigert, R.; Chavakis, T.; Adams, R.H.; et al. RhoA and ROCK Mediate Histamine-Induced Vascular Leakage and Anaphylactic Shock. Nat. Commun. 2015, 6, 6725. [Google Scholar] [CrossRef]
- Pronk, M.C.A.; van Bezu, J.S.M.; van Nieuw Amerongen, G.P.; van Hinsbergh, V.W.M.; Hordijk, P.L. RhoA, RhoB and RhoC Differentially Regulate Endothelial Barrier Function. Small GTPases 2017, 10, 466–484. [Google Scholar] [CrossRef]
- van der Krogt, J.M.; van der Meulen, I.J.; van Buul, J.D. Spatiotemporal Regulation of Rho GTPase Signaling during Endothelial Barrier Remodeling. Curr. Opin. Physiol. 2023, 34, 100676. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.Y.; Cartwright, C.A. Regulation of the Src Tyrosine Kinase and Syp Tyrosine Phosphatase by Their Cellular Association. Oncogene 1995, 11, 1955–1962. [Google Scholar] [PubMed]
- Sausgruber, N.; Coissieux, M.-M.; Britschgi, A.; Wyckoff, J.; Aceto, N.; Leroy, C.; Stadler, M.B.; Voshol, H.; Bonenfant, D.; Bentires-Alj, M. Tyrosine Phosphatase SHP2 Increases Cell Motility in Triple-Negative Breast Cancer through the Activation of SRC-Family Kinases. Oncogene 2015, 34, 2272–2278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Q.; Yang, W.; Kontaridis, M.I.; Bivona, T.G.; Wen, G.; Araki, T.; Luo, J.; Thompson, J.A.; Schraven, B.L.; Philips, M.R.; et al. Shp2 Regulates Src Family Kinase Activity and Ras/Erk Activation by Controlling Csk Recruitment. Mol. Cell 2004, 13, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Meng, S.; Mei, L.; Zhao, Z.J.; Jove, R.; Wu, J. Roles of Gab1 and SHP2 in Paxillin Tyrosine Dephosphorylation and Src Activation in Response to Epidermal Growth Factor. J. Biol. Chem. 2004, 279, 8497–8505. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J. Endothelial Permeability and VE-Cadherin: A Wacky Comradeship. Cell Adhes. Migr. 2013, 7, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Vilgrain, I.; Sidibé, A.; Polena, H.; Cand, F.; Mannic, T.; Arboleas, M.; Boccard, S.; Baudet, A.; Gulino-Debrac, D.; Bouillet, L.; et al. Evidence for Post-Translational Processing of Vascular Endothelial (VE)-Cadherin in Brain Tumors: Towards a Candidate Biomarker. PLoS ONE 2013, 8, e80056. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-S.; Manser, E. PAK Family Kinases: Physiological Roles and Regulation. Cell Logist. 2012, 2, 59–68. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F. Endothelial Adherens Junctions at a Glance. J. Cell Sci. 2013, 126, 2545–2549. [Google Scholar] [CrossRef]
- Adam, A.P. Regulation of Endothelial Adherens Junctions by Tyrosine Phosphorylation. Mediat. Inflamm. 2015, 2015, 272858. [Google Scholar] [CrossRef]
- Miner, J.J.; Daniels, B.P.; Shrestha, B.; Proenca-Modena, J.L.; Lew, E.D.; Lazear, H.M.; Gorman, M.J.; Lemke, G.; Klein, R.S.; Diamond, M.S. The TAM Receptor Mertk Protects against Neuroinvasive Viral Infection by Maintaining Blood-Brain Barrier Integrity. Nat. Med. 2015, 21, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Lippoldt, A. Tight Junctions of the Blood–Brain Barrier: Development, Composition and Regulation. Vascul. Pharmacol. 2002, 15, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Rochefort, P.; Chabaud, S.; Pierga, J.-Y.; Tredan, O.; Brain, E.; Bidard, F.-C.; Schiffler, C.; Polena, H.; Khalil-Mgharbel, A.; Vilgrain, I.; et al. Soluble VE-Cadherin in Metastatic Breast Cancer: An Independent Prognostic Factor for Both Progression-Free Survival and Overall Survival. Br. J. Cancer 2017, 116, 356–361. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Name/Catalog# | Supplier | Type of Antibody | Concentration |
|---|---|---|---|---|
| Primary antibodies | ||||
| Actin | β-actin (13E5) HRP conjugate/#5125 | Cell Signaling Technology (Danvers, MA, USA) | Monoclonal Rabbit IgG | WB 1:1000 |
| HSP27 | HSP27 (D6W5V)/#95357 | Cell Signaling Technology | Monoclonal Rabbit IgG | WB 1:1000 |
| p-HSP27 | Phospho-HSP27 (Ser82)/#2401 | Cell Signaling Technology | Polyclonal Rabbit AB | WB 1:1000 |
| MLC | Myosin Light Chain 2 (D18E2)/#8505 | Cell Signaling Technology | Monoclonal Rabbit IgG | WB 1:1000 |
| p-MLC | Phospho-Myosin Light Chain 2 (Thr18/Ser19)/#3674 | Cell Signaling Technology | Polyclonal Rabbit AB | WB 1:1000 |
| PAK | PAK1 Antibody/#2602 | Cell Signaling Technology | Polyclonal Rabbit AB | WB 1:1000 |
| p-PAK | Phospho-PAK1(Ser144)/PAK2 (Ser141)/#2606 | Cell Signaling Technology | Polyclonal Rabbit AB | WB 1:1000 |
| c-Src | Src Antibody/#2108 | Cell Signaling Technology | Polyclonal Rabbit AB | WB 1:1000 |
| p-c-Src | Phospho-Src Family (Tyr416)/#2101 | Cell Signaling Technology | Polyclonal Rabbit AB | WB 1:1000 |
| VEC | Purified Mouse Anti-Human CD144 (55-7H1)/#555661 | BD Biosciences | Monoclonal Mouse IgG | WB 1:1000 IF 1:400 |
| Anti-VE-cadherin AB, (clone BV6) /#MABT134 | Millipore | Monoclonal Mouse IgG | WB 1:1000 | |
| p-VEC | Phospho-VEC (Ser665) | * | Polyclonal Rabbit AB | WB 1:1000 |
| Phospho-VEC (Tyr 685) | ** | Polyclonal Rabbit AB | WB 1:1000 | |
| Secondary antibodies | ||||
| - | Anti-Mouse IgG—Peroxidase AB /#A4416 | Sigma-Aldrich (Burlington, MA, USA) | Polyclonal Goat IgG | WB 1:4000/1:5000 |
| - | Anti-Rabbit IgG—Peroxidase AB /#A0545 | Sigma-Aldrich | Polyclonal Goat IgG | WB 1:4000/1:5000 |
| - | Anti-Mouse/Rabbit IgG Antibody (H+L), Biotinylated/#BA-1400 | Vector Laboratories | Polyclonal Horse IgG | IF 1:100 |
| Neutralizing and Irrelevant antibodies | ||||
| AXL | Human AXL Antibody/#AF154 | R&D Systems (Minneapolis, MN, USA) | Polyclonal Goat IgG | 10 µg/mL |
| MERTK | Human MERTK Antibody/#AF891 | R&D Systems | Polyclonal Goat IgG | 10 µg/mL |
| PROS1 | Anti-Human Protein S /#A038401-2 | Agilent Dako (Santa Clara, CA, USA) | Polyclonal Rabbit AB | 10 µg/mL |
| TYRO3 | Human Dtk Antibody/#AF859 | R&D Systems | Polyclonal Goat IgG | 10 µg/mL |
| - | Normal Rabbit IgG Control/#AB105C | R&D Systems | Polyclonal Rabbit IgG | 10 µg/mL |
| - | Normal Goat IgG Control/#AB108C | R&D Systems | Polyclonal Goat IgG | 10 µg/mL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joussaume, A.; Kanthou, C.; Pardo, O.E.; Karayan-Tapon, L.; Benzakour, O.; Dkhissi, F. The Vitamin K-Dependent Anticoagulant Factor, Protein S, Regulates Vascular Permeability. Curr. Issues Mol. Biol. 2024, 46, 3278-3293. https://doi.org/10.3390/cimb46040205
Joussaume A, Kanthou C, Pardo OE, Karayan-Tapon L, Benzakour O, Dkhissi F. The Vitamin K-Dependent Anticoagulant Factor, Protein S, Regulates Vascular Permeability. Current Issues in Molecular Biology. 2024; 46(4):3278-3293. https://doi.org/10.3390/cimb46040205
Chicago/Turabian StyleJoussaume, Aurélie, Chryso Kanthou, Olivier E. Pardo, Lucie Karayan-Tapon, Omar Benzakour, and Fatima Dkhissi. 2024. "The Vitamin K-Dependent Anticoagulant Factor, Protein S, Regulates Vascular Permeability" Current Issues in Molecular Biology 46, no. 4: 3278-3293. https://doi.org/10.3390/cimb46040205
APA StyleJoussaume, A., Kanthou, C., Pardo, O. E., Karayan-Tapon, L., Benzakour, O., & Dkhissi, F. (2024). The Vitamin K-Dependent Anticoagulant Factor, Protein S, Regulates Vascular Permeability. Current Issues in Molecular Biology, 46(4), 3278-3293. https://doi.org/10.3390/cimb46040205