
Single-Cell Sequencing Technology in Ruminant Livestock: Challenges and Opportunities

Abstract
1. Introduction
2. Brief Overview of Single-Cell and Single-Nuclei mRNA and ATAC Sequencing
2.1. Cell Isolation, Library Preparation, and Sequencing
2.2. Bioinformatic Analyses
2.3. Data Interpretation
3. Current Uses of Single-Cell Sequencing in Ruminant Livestock Research
3.1. Cellular Heterogeneity within Tissues Important for Food and Fiber Production
3.2. Gene Expression Dynamics during Reproduction and Development at Single-Cell Resolution
3.3. Immune Cell Gene Expression and Chromatin Profiling
4. Challenges and Limitations of Single-Cell Technologies in Ruminant Livestock Research
4.1. Technical and Bioinformatic Challenges
4.2. Cell Cluster Identification and Spatial Resolution
4.3. Reproducibility, Collaboration, and Knowledge Sharing
5. The Future of Single-Cell Sequencing in Ruminant Livestock Research
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sun, S.; Shen, X.; Li, Y.; Wang, S.; Li, R.; Zhang, H.; Shen, G.; Guo, B.; Wei, J.; Xu, J.; et al. Single-cell RNA sequencing provides a high-resolution roadmap for understanding the multicellular compartmentation of specialized metabolism. Nat. Plants 2023, 9, 179–190. [Google Scholar] [CrossRef]
- Ke, M.; Elshenawy, B.; Sheldon, H.; Arora, A.; Buffa, F.M. Single cell RNA-sequencing: A powerful yet still challenging technology to study cellular heterogeneity. Bioessays 2022, 44, e2200084. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Park, C.; Kim, M.; Kim, H.; Kim, J.; Lee, D.S. Advances in single-cell omics and multiomics for high-resolution molecular profiling. Exp. Mol. Med. 2024, 56, 515–526. [Google Scholar] [CrossRef]
- Ogbeide, S.; Giannese, F.; Mincarelli, L.; Macaulay, I.C. Into the multiverse: Advances in single-cell multiomic profiling. Trends Genet. 2022, 38, 831–843. [Google Scholar] [CrossRef]
- Baysoy, A.; Bai, Z.; Satija, R.; Fan, R. The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 2023, 24, 695–713. [Google Scholar] [CrossRef]
- Wu, J.J.; Zhu, S.; Gu, F.; Valencak, T.G.; Liu, J.X.; Sun, H.Z. Cross-tissue single-cell transcriptomic landscape reveals the key cell subtypes and their potential roles in the nutrient absorption and metabolism in dairy cattle. J. Adv. Res. 2022, 37, 1–18. [Google Scholar] [CrossRef]
- Michelotti, T.C.; Kisby, B.R.; Flores, L.S.; Tegeler, A.P.; Fokar, M.; Crasto, C.; Menarim, B.C.; Loux, S.C.; Strieder-Barboza, C. Single-nuclei analysis reveals depot-specific transcriptional heterogeneity and depot-specific cell types in adipose tissue of dairy cows. Front. Cell Dev. Biol. 2022, 10, 1025240. [Google Scholar] [CrossRef]
- Lavagi, I.; Krebs, S.; Simmet, K.; Beck, A.; Zakhartchenko, V.; Wolf, E.; Blum, H. Single-cell RNA sequencing reveals developmental heterogeneity of blastomeres during major genome activation in bovine embryos. Sci. Rep. 2018, 8, 4071. [Google Scholar] [CrossRef] [PubMed]
- Scatolin, G.N.; Ming, H.; Wang, Y.; Iyyappan, R.; Gutierrez-Castillo, E.; Zhu, L.; Sagheer, M.; Song, C.; Bondioli, K.; Jiang, Z. Single-cell transcriptional landscapes of bovine peri-implantation development. iScience 2024, 27, 109605. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Long, C.; Zhao, G.; Su, J.; Ren, J.; Sun, W.; Wang, Z.; Zhang, J.; Liu, M.; Hao, C.; et al. Reprogramming barriers in bovine cells nuclear transfer revealed by single-cell RNA-seq analysis. J. Cell Mol. Med. 2022, 26, 4792–4804. [Google Scholar] [CrossRef]
- Ortega, M.S.; Rizo, J.A.; Drum, J.N.; O‘Neil, E.V.; Pohler, K.G.; Kerns, K.; Schmelze, A.; Green, J.; Spencer, T.E. Development of an Improved in vitro Model of Bovine Trophectoderm Differentiation. Front. Anim. Sci. 2022, 3, 898808. [Google Scholar] [CrossRef]
- Soto, D.A.; Ross, P.J. Similarities between bovine and human germline development revealed by single-cell RNA sequencing. Reproduction 2021, 161, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Zorc, M.; Dolinar, M.; Dovč, P. A Single-Cell Transcriptome of Bovine Milk Somatic Cells. Genes 2024, 15, 349. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, P.; Li, C.; Liu, Q.; Yao, Z.; Li, Y.; Zhang, X.; Sun, J.; Simintiras, C.; Welborn, M.; et al. A single-cell atlas of bovine skeletal muscle reveals mechanisms regulating intramuscular adipogenesis and fibrogenesis. J. Cachexia Sarcopenia Muscle 2023, 14, 2152–2167. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Wan, P.; Wang, H.; Cai, X.; Wang, J.; Chai, Z.; Zhang, M.; Yang, N.; Wu, Z.; Zhu, J.; et al. Transcriptional and open chromatin analysis of bovine skeletal muscle development by single-cell sequencing. Cell Prolif. 2023, 56, e13430. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Y.; Li, C.J.; Fang, L.; Liu, G.E.; Zhao, X.; Zhang, Y.; Cai, G.; Xue, G.; Liu, Y.; et al. The single-cell transcriptome and chromatin accessibility datasets of peripheral blood mononuclear cells in Chinese holstein cattle. BMC Genom. Data 2023, 24, 39. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, J.; Cai, G.; Wang, Y.; Yang, W.; Li, Y.; Zhao, X.; Li, R.; Tuo, W.; Baldwin, R.L.; et al. Single-cell transcriptomic and chromatin accessibility analyses of dairy cattle peripheral blood mononuclear cells and their responses to lipopolysaccharide. BMC Genom. 2022, 23, 338. [Google Scholar] [CrossRef]
- Davenport, K.M.; O‘Neil, E.V.; Ortega, M.S.; Patterson, A.; Kelleher, A.M.; Warren, W.C.; Spencer, T.E. Single-cell insights into development of the bovine placenta. Biol. Reprod. 2024, 110, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Davenport, K.M.; Ortega, M.S.; Liu, H.; O’Neil, E.V.; Kelleher, A.M.; Warren, W.C.; Spencer, T.E. Single-nuclei RNA sequencing (snRNA-seq) uncovers trophoblast cell types and lineages in the mature bovine placenta. Proc. Natl. Acad. Sci. USA 2023, 120, e2221526120. [Google Scholar] [CrossRef]
- Becker, D.; Weikard, R.; Hadlich, F.; Kühn, C. Single-cell RNA sequencing of freshly isolated bovine milk cells and cultured primary mammary epithelial cells. Sci. Data 2021, 8, 177. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, L.; Baldwin, R.L.; Connor, E.E.; Cole, J.B.; Van Tassell, C.P.; Ma, L.; Li, C.J.; Liu, G.E. Single-cell transcriptomic analyses of dairy cattle ruminal epithelial cells during weaning. Genomics 2021, 113, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Lyu, P.; Qi, Y.; Tu, Z.J.; Jiang, H. Single-cell RNA Sequencing Reveals Heterogeneity of Cultured Bovine Satellite Cells. Front. Genet. 2021, 12, 742077. [Google Scholar] [CrossRef] [PubMed]
- Messmer, T.; Dohmen, R.G.J.; Schaeken, L.; Melzener, L.; Hueber, R.; Godec, M.; Didoss, C.; Post, M.J.; Flack, J.E. Single-cell analysis of bovine muscle-derived cell types for cultured meat production. Front. Nutr. 2023, 10, 1212196. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.X.; Ma, W.J.; Wu, Z.B.; Li, S.; Zhang, X.Q.; He, Z.; Wu, S.X.; Tao, H.P.; Fang, Y.; Song, Y.W.; et al. Single-cell transcriptomic characterization of sheep conceptus elongation and implantation. Cell Rep. 2023, 42, 112860. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Guo, W.; Yang, G.; Su, H.; Dou, A.; Chen, L.; Ma, T.; Su, J.; Liu, M.; Su, B.; et al. A Single-Cell Atlas of an Early Mongolian Sheep Embryo. Vet. Sci. 2023, 10, 543. [Google Scholar] [CrossRef] [PubMed]
- Ge, T.; Wen, Y.; Li, B.; Huang, X.; Jiang, S.; Zhang, E. Single-cell sequencing reveals the reproductive variations between primiparous and multiparous Hu ewes. J. Anim. Sci. Biotechnol. 2023, 14, 144. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Sun, D.M.; Qin, T.; Mao, S.Y.; Zhu, W.Y.; Yin, Y.Y.; Huang, J.; Heller, R.; Li, Z.P.; Liu, J.H.; et al. Single-cell transcriptomic landscape of the sheep rumen provides insights into physiological programming development and adaptation of digestive strategies. Zool. Res. 2022, 43, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Liu, Y.J.; Wei, W.T.; Huang, Q.X.; Zhao, L.P.; Luo, L.Y.; Zhu, Q.; Zhang, L.; Chen, Y.; Ren, Y.L.; et al. Single-cell transcriptome and metagenome profiling reveals the genetic basis of rumen functions and convergent developmental patterns in ruminants. Genome Res. 2023, 33, 1690–1707. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wu, T.; Sun, J.; Li, Y.; Yuan, Z.; Sun, W. Single-Cell Transcriptomics Reveals the Molecular Anatomy of Sheep Hair Follicle Heterogeneity and Wool Curvature. Front. Cell Dev. Biol. 2021, 9, 800157. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, T.; Li, J.; Niu, C.; Sun, W.; Zhu, S.; Zhao, H.; Qiao, G.; Han, M.; He, X.; et al. The Transcriptional Cell Atlas of Testis Development in Sheep at Pre-Sexual Maturity. Curr. Issues Mol. Biol. 2022, 44, 483–497. [Google Scholar] [CrossRef]
- Tian, Y.; Sun, P.; Liu, W.X.; Shan, L.Y.; Hu, Y.T.; Fan, H.T.; Shen, W.; Liu, Y.B.; Zhou, Y.; Zhang, T. Single-cell RNA sequencing of the Mongolia sheep testis reveals a conserved and divergent transcriptome landscape of mammalian spermatogenesis. FASEB J. 2022, 36, e22348. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Long, X.; Hao, F.; Kang, J.; Wang, N.; Wang, Y.; Wang, M.; Gao, Y.; Zhou, M.; Duo, L.; et al. Integration of single-cell transcriptome and chromatin accessibility of early gonads development among goats, pigs, macaques, and humans. Cell Rep. 2022, 41, 111587. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Sun, J.; Zhu, J.; Ding, X.; Lan, T.; Wang, X.; Wu, W.; Ou, Z.; Zhu, L.; Ding, P.; et al. Single cell atlas for 11 non-model mammals, reptiles and birds. Nat. Commun. 2021, 12, 7083. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Zhao, Y.; Ma, D.; Zhao, M.; Li, N.; Men, Y.; Zhang, Y.; Chu, H.; Lei, C.; et al. scRNA-seq of ovarian follicle granulosa cells from different fertility goats reveals distinct expression patterns. Reprod. Domest. Anim. 2021, 56, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Song, S.; Wang, F.; Li, Y.; Li, Z.; Yao, H.; Zhao, Y.; Zhao, Z. Single-cell transcriptomic atlas of goat ovarian aging. J. Anim. Sci. Biotechnol. 2023, 14, 151. [Google Scholar] [CrossRef]
- Ge, W.; Zhang, W.; Zhang, Y.; Zheng, Y.; Li, F.; Wang, S.; Liu, J.; Tan, S.; Yan, Z.; Wang, L.; et al. A Single-cell Transcriptome Atlas of Cashmere Goat Hair Follicle Morphogenesis. Genom. Proteom. Bioinform. 2021, 19, 437–451. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Hui, T.; Chen, R.; Xu, Y.; Zhang, Y.; Tian, H.; Wang, W.; Cong, Y.; Guo, S.; et al. Single-Cell Sequencing Reveals Differential Cell Types in Skin Tissues of Liaoning Cashmere Goats and Key Genes Related Potentially to the Fineness of Cashmere Fiber. Front. Genet. 2021, 12, 726670. [Google Scholar] [CrossRef]
- Yang, F.; Li, R.; Zhao, C.; Che, T.; Guo, J.; Xie, Y.; Wang, Z.; Li, J.; Liu, Z. Single-cell sequencing reveals the new existence form of dermal papilla cells in the hair follicle regeneration of cashmere goats. Genomics 2022, 114, 110316. [Google Scholar] [CrossRef]
- Chen, M.; Wang, N.; Yang, H.; Liu, D.; Gao, Y.; Duo, L.; Cui, X.; Hao, F.; Ye, J.; Gao, F.; et al. Single-cell transcriptome analysis of the germ cells and somatic cells during mitotic quiescence stage in goats. FASEB J. 2023, 37, e23244. [Google Scholar] [CrossRef]
- Sargison, N.D. The critical importance of planned small ruminant livestock health and production in addressing global challenges surrounding food production and poverty alleviation. N. Z. Vet. J. 2020, 68, 136–144. [Google Scholar] [CrossRef]
- Davis, T.C.; White, R.R. Breeding animals to feed people: The many roles of animal reproduction in ensuring global food security. Theriogenology 2020, 150, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, A.S.E.; Nathan, A.; Raychaudhuri, S.; MacArthur, D.G.; Powell, J.E. Single-cell genomics meets human genetics. Nat. Rev. Genet. 2023, 24, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Vandereyken, K.; Sifrim, A.; Thienpont, B.; Voet, T. Methods and applications for single-cell and spatial multi-omics. Nat. Rev. Genet. 2023, 24, 494–515. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, J.M.; Wang, G.; Jonika, M.M.; Gill, C.A.; Blackmon, H.; Athrey, G.N. A Primer for Single-Cell Sequencing in Non-Model Organisms. Genes 2022, 13, 380. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.I.; Fuentes-Trillo, A.; Domínguez Conde, C. Opportunities and tradeoffs in single-cell transcriptomic technologies. Trends Genet. 2024, 40, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.L.; Archibald, A.L.; Daetwyler, H.D.; Groenen, M.A.M.; Harrison, P.W.; Houston, R.D.; Kühn, C.; Lien, S.; Macqueen, D.J.; Reecy, J.M.; et al. From FAANG to fork: Application of highly annotated genomes to improve farmed animal production. Genome Biol. 2020, 21, 285. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.W.; Sokolov, A.; Nayak, A.; Fan, J.; Zerbino, D.; Cochrane, G.; Flicek, P. The FAANG Data Portal: Global, Open-Access, “FAIR”, and Richly Validated Genotype to Phenotype Data for High-Quality Functional Annotation of Animal Genomes. Front. Genet. 2021, 12, 639238. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.P.L.; Bickhart, D.M.; Boichard, D.; Chamberlain, A.J.; Djikeng, A.; Jiang, Y.; Low, W.Y.; Pausch, H.; Demyda-Peyrás, S.; Prendergast, J.; et al. The Bovine Pangenome Consortium: Democratizing production and accessibility of genome assemblies for global cattle breeds and other bovine species. Genome Biol. 2023, 24, 139. [Google Scholar] [CrossRef]
- Woolley, S.A.; Salavati, M.; Clark, E.L. Recent advances in the genomic resources for sheep. Mamm. Genome 2023, 34, 545–558. [Google Scholar] [CrossRef]
- Jovic, D.; Liang, X.; Zeng, H.; Lin, L.; Xu, F.; Luo, Y. Single-cell RNA sequencing technologies and applications: A brief overview. Clin. Transl. Med. 2022, 12, e694. [Google Scholar] [CrossRef]
- Yao, C.; Bora, S.A.; Chen, P.; Goodridge, H.S.; Gharib, S.A. Sample processing and single cell RNA-sequencing of peripheral blood immune cells from COVID-19 patients. STAR Protoc. 2021, 2, 100582. [Google Scholar] [CrossRef] [PubMed]
- Petrany, M.J.; Swoboda, C.O.; Sun, C.; Chetal, K.; Chen, X.; Weirauch, M.T.; Salomonis, N.; Millay, D.P. Single-nucleus RNA-seq identifies transcriptional heterogeneity in multinucleated skeletal myofibers. Nat. Commun. 2020, 11, 6374. [Google Scholar] [CrossRef]
- Sun, W.; Dong, H.; Balaz, M.; Slyper, M.; Drokhlyansky, E.; Colleluori, G.; Giordano, A.; Kovanicova, Z.; Stefanicka, P.; Balazova, L.; et al. snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature 2020, 587, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kang, H.; Jo, A.; Yoo, S.A.; Lee, H.O. Perspectives on single-nucleus RNA sequencing in different cell types and tissues. J. Pathol. Transl. Med. 2023, 57, 52–59. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Shi, Z.; Das, S.; Morabito, S.; Miyoshi, E.; Swarup, V. Protocol for single-nucleus ATAC sequencing and bioinformatic analysis in frozen human brain tissue. STAR Protoc. 2022, 3, 101491. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, L.; Mohammed Ismail, W.; Mazzone, A.; Dumbrava, M.; Fernandez, J.; Munankarmy, A.; Lasho, T.; Binder, M.; Simon, V.; Kim, K.H.; et al. Characterization and Optimization of Multiomic Single-Cell Epigenomic Profiling. Genes 2023, 14, 1245. [Google Scholar] [CrossRef]
- Heumos, L.; Schaar, A.C.; Lance, C.; Litinetskaya, A.; Drost, F.; Zappia, L.; Lücken, M.D.; Strobl, D.C.; Henao, J.; Curion, F.; et al. Best practices for single-cell analysis across modalities. Nat. Rev. Genet. 2023, 24, 550–572. [Google Scholar] [CrossRef]
- Yang, S.; Corbett, S.E.; Koga, Y.; Wang, Z.; Johnson, W.E.; Yajima, M.; Campbell, J.D. Decontamination of ambient RNA in single-cell RNA-seq with DecontX. Genome Biol. 2020, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.J.; Chaffin, M.D.; Arduini, A.; Akkad, A.D.; Banks, E.; Marioni, J.C.; Philippakis, A.A.; Ellinor, P.T.; Babadi, M. Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. Nat. Methods 2023, 20, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Amezquita, R.A.; Lun, A.T.L.; Becht, E.; Carey, V.J.; Carpp, L.N.; Geistlinger, L.; Marini, F.; Rue-Albrecht, K.; Risso, D.; Soneson, C.; et al. Orchestrating single-cell analysis with Bioconductor. Nat. Methods 2020, 17, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [PubMed]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Spielmann, M.; Qiu, X.; Huang, X.; Ibrahim, D.M.; Hill, A.J.; Zhang, F.; Mundlos, S.; Christiansen, L.; Steemers, F.J.; et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Van den Berge, K.; Roux de Bézieux, H.; Street, K.; Saelens, W.; Cannoodt, R.; Saeys, Y.; Dudoit, S.; Clement, L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 2020, 11, 1201. [Google Scholar] [CrossRef] [PubMed]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef]
- Zheng, S.C.; Stein-O’Brien, G.; Boukas, L.; Goff, L.A.; Hansen, K.D. Pumping the brakes on RNA velocity by understanding and interpreting RNA velocity estimates. Genome Biol. 2023, 24, 246. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef]
- Wang, S.; Zheng, H.; Choi, J.S.; Lee, J.K.; Li, X.; Hu, H. A systematic evaluation of the computational tools for ligand-receptor-based cell-cell interaction inference. Brief. Funct. Genom. 2022, 21, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Liu, H.; Yu, J.; Kelliher, M.A.; Castilla, L.H.; Lawson, N.D.; Zhu, L.J. ATACseqQC: A Bioconductor package for post-alignment quality assessment of ATAC-seq data. BMC Genom. 2018, 19, 169. [Google Scholar] [CrossRef] [PubMed]
- Germain, P.L.; Lun, A.; Garcia Meixide, C.; Macnair, W.; Robinson, M.D. Doublet identification in single-cell sequencing data using. F1000Research 2021, 10, 979. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lareau, C.; Andreani, T.; Vinyard, M.E.; Garcia, S.P.; Clement, K.; Andrade-Navarro, M.A.; Buenrostro, J.D.; Pinello, L. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 2019, 20, 241. [Google Scholar] [CrossRef] [PubMed]
- Granja, J.M.; Corces, M.R.; Pierce, S.E.; Bagdatli, S.T.; Choudhry, H.; Chang, H.Y.; Greenleaf, W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021, 53, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Srivastava, A.; Madad, S.; Lareau, C.A.; Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 2021, 18, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, C.; Quintero, A.; Wu, L.; Yuan, Y.; Wang, M.; Cheng, M.; Leng, L.; Xu, L.; Dong, G.; et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat. Commun. 2019, 10, 470. [Google Scholar] [CrossRef] [PubMed]
- Kartha, V.K.; Duarte, F.M.; Hu, Y.; Ma, S.; Chew, J.G.; Lareau, C.A.; Earl, A.; Burkett, Z.D.; Kohlway, A.S.; Lebofsky, R.; et al. Functional inference of gene regulation using single-cell multi-omics. Cell Genom. 2022, 2, 100166. [Google Scholar] [CrossRef]
- Fleck, J.S.; Jansen, S.M.J.; Wollny, D.; Zenk, F.; Seimiya, M.; Jain, A.; Okamoto, R.; Santel, M.; He, Z.; Camp, J.G.; et al. Inferring and perturbing cell fate regulomes in human brain organoids. Nature 2023, 621, 365–372. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lonnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genom. Med. 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.C.; Townes, F.W.; Teng, M.; Irizarry, R.A. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 2018, 19, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Squair, J.W.; Courtine, G. Enabling reproducible re-analysis of single-cell data. Genome Biol. 2021, 22, 215. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Triant, D.A.; Walsh, A.T.; Hartley, G.A.; Petry, B.; Stegemiller, M.R.; Nelson, B.M.; McKendrick, M.M.; Fuller, E.P.; Cockett, N.E.; Koltes, J.E.; et al. AgAnimalGenomes: Browsers for viewing and manually annotating farm animal genomes. Mamm. Genome 2023, 34, 418–436. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Franzén, O.; Gan, L.M.; Björkegren, J.L.M. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 2022, 13, 1246. [Google Scholar] [CrossRef]
- Galdos, F.X.; Xu, S.; Goodyer, W.R.; Duan, L.; Huang, Y.V.; Lee, S.; Zhu, H.; Lee, C.; Wei, N.; Lee, D.; et al. devCellPy is a machine learning-enabled pipeline for automated annotation of complex multilayered single-cell transcriptomic data. Nat. Commun. 2022, 13, 5271. [Google Scholar] [CrossRef]
- Hou, W.; Ji, Z. Assessing GPT-4 for cell type annotation in single-cell RNA-seq analysis. Nat. Methods 2024. [CrossRef]
- Adema, K.; Schon, M.A.; Nodine, M.D.; Kohlen, W. Lost in space: What single-cell RNA sequencing cannot tell you. Trends Plant Sci. 2024, in press. [CrossRef]
- Spencer, T.E.; Lowke, M.T.; Davenport, K.M.; Kelleher, A.M. Single-cell insights into epithelial morphogenesis in the neonatal mouse uterus. Proc. Natl. Acad. Sci. USA 2023, 120, e2316410120. [Google Scholar] [CrossRef]
- Marx, V. Publisher Correction: Method of the Year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 219. [Google Scholar] [CrossRef]
- Williams, C.G.; Lee, H.J.; Asatsuma, T.; Vento-Tormo, R.; Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 2022, 14, 68. [Google Scholar] [CrossRef]
- Phan, Q.M.; Driskell, I.M.; Driskell, R.R. The Three Rs of Single-Cell RNA Sequencing: Reuse, Refine, and Resource. J. Investig. Dermatol. 2021, 141, 1627–1629. [Google Scholar] [CrossRef]
- Grones, C.; Eekhout, T.; Shi, D.; Neumann, M.; Berg, L.S.; Ke, Y.; Shahan, R.; Cox, K.L.; Gomez-Cano, F.; Nelissen, H.; et al. Best practices for the execution, analysis, and data storage of plant single-cell/nucleus transcriptomics. Plant Cell 2024, 36, 812–828. [Google Scholar] [CrossRef] [PubMed]
- Shafer, M.E.R. Cross-Species Analysis of Single-Cell Transcriptomic Data. Front. Cell Dev. Biol. 2019, 7, 175. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Miao, Z.; Brazma, A.; Papatheodorou, I. Benchmarking strategies for cross-species integration of single-cell RNA sequencing data. Nat. Commun. 2023, 14, 6495. [Google Scholar] [CrossRef] [PubMed]
- Jiao, A.; Zhang, C.; Wang, X.; Sun, L.; Liu, H.; Su, Y.; Lei, L.; Li, W.; Ding, R.; Ding, C.; et al. Single-cell sequencing reveals the evolution of immune molecules across multiple vertebrate species. J. Adv. Res. 2024, 55, 73–87. [Google Scholar] [CrossRef]
- Tang, X.; Huang, Y.; Lei, J.; Luo, H.; Zhu, X. The single-cell sequencing: New developments and medical applications. Cell Biosci. 2019, 9, 53. [Google Scholar] [CrossRef]
- Fahlgren, N.; Kapoor, M.; Yordanova, G.; Papatheodorou, I.; Waese, J.; Cole, B.; Harrison, P.; Ware, D.; Tickle, T.; Paten, B.; et al. Toward a data infrastructure for the Plant Cell Atlas. Plant Physiol. 2023, 191, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Uribe, J.; Wiarda, J.E.; Sivasankaran, S.K.; Daharsh, L.; Liu, H.; Byrne, K.A.; Smith, T.P.L.; Lunney, J.K.; Loving, C.L.; Tuggle, C.K. Reference Transcriptomes of Porcine Peripheral Immune Cells Created Through Bulk and Single-Cell RNA Sequencing. Front. Genet. 2021, 12, 689406. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.X.; Chen, Z.C.; Zhong, J.Y.; Hu, K.H.; Zheng, Y.F.; Chen, Y.; Xie, S.Q.; Bo, X.C.; Luo, F.; Tang, C.; et al. High-throughput and high-accuracy single-cell RNA isoform analysis using PacBio circular consensus sequencing. Nat. Commun. 2023, 14, 2631. [Google Scholar] [CrossRef] [PubMed]
- Bartosovic, M.; Kabbe, M.; Castelo-Branco, G. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 2021, 39, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhou, T.; Ma, J. Multiscale and integrative single-cell Hi-C analysis with Higashi. Nat. Biotechnol. 2022, 40, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Sun, J.; Zhao, C.; Liu, X.; Zhang, X.; Jiang, S.; Wei, C.; Yu, H.; Zeng, X.; Fan, L.; et al. Simultaneous profiling of chromatin architecture and transcription in single cells. Nat. Struct. Mol. Biol. 2023, 30, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.V.; O‘Connell, B.L.; Mulqueen, R.M.; Thomas, J.; Woodfin, A.R.; Acharya, S.; Mandel, G.; Pokholok, D.; Steemers, F.J.; Adey, A.C. High-throughput robust single-cell DNA methylation profiling with sciMETv2. Nat. Commun. 2022, 13, 7627. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zeng, Q.; Zhou, J.; Bartlett, A.; Wang, B.A.; Berube, P.; Tian, W.; Kenworthy, M.; Altshul, J.; Nery, J.R.; et al. Single-cell DNA methylome and 3D multi-omic atlas of the adult mouse brain. Nature 2023, 624, 366–377. [Google Scholar] [CrossRef]
- Tuggle, C.K.; Clarke, J.L.; Murdoch, B.M.; Lyons, E.; Scott, N.M.; Beneš, B.; Campbell, J.D.; Chung, H.; Daigle, C.L.; Das Choudhury, S.; et al. Current challenges and future of agricultural genomes to phenomes in the USA. Genome Biol. 2024, 25, 8. [Google Scholar] [CrossRef]

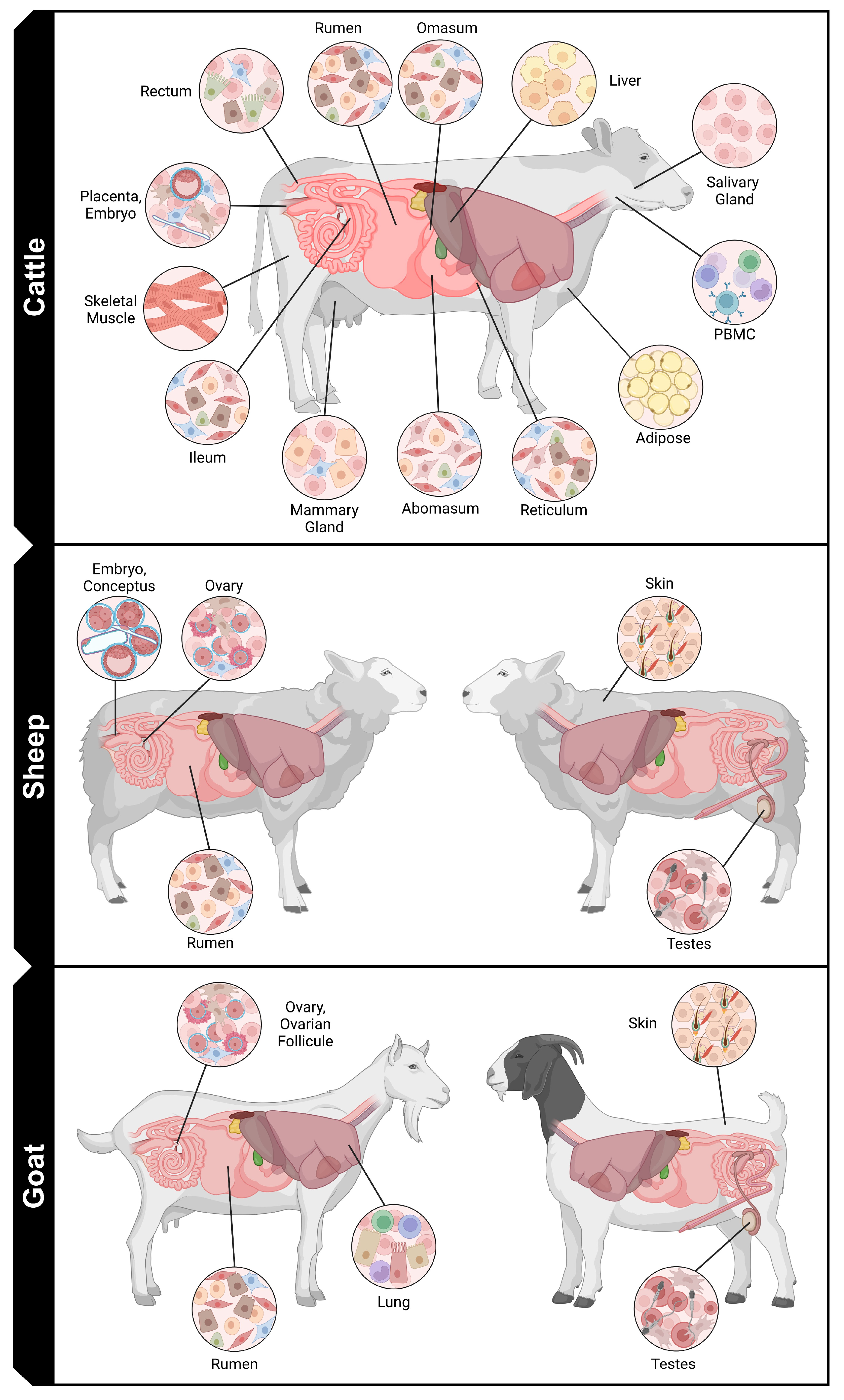

{kind=link}
{kind=link}
| Tissue(s) or Cell Type(s) | Isolation Type | Data Type | Database and Accession * | Reference |
|---|---|---|---|---|
| Abomasum, ileum, liver, mammary gland, omasum, PBMC *, rectum, reticulum, rumen, salivary gland | Cell | RNA | GEO GSE176512 | [6] |
| Adipose | Nuclei | RNA | GEO GSE211707 | [7] |
| Embryo (8- and 16-cell stage) | Cell | RNA | GEO GSE99210 | [8] |
| Embryo (peri-implantation) | Cell | RNA | GEO GSE234335 | [9] |
| Embryo (SCNT) | Cell | RNA | SRA PRJNA727165 | [10] |
| Embryo (trophectoderm) | Cell | RNA | GEO GSE200216 | [11] |
| Fetal Gonads | Cell | RNA | GEO GSE162952 | [12] |
| Milk Somatic Cells | Cell | RNA | ENA PRJEB73560 | [13] |
| Muscle | Cell | RNA | GEO GSE205347 | [14] |
| Muscle | Cell | RNA, ATAC | GSA CRA006626 | [15] |
| PBMC * | Cell | RNA, ATAC | GEO GSE225962 | [16] |
| PBMC * | Cell | RNA, ATAC | GEO GSE166473 | [17] |
| Placenta (developing) | Cell | RNA | GEO GSE234524 | [18] |
| Placenta (mature) | Nuclei | RNA | GEO GSE214407 | [19] |
| Primary Mammary Epithelial Cells | Cell | RNA | FAANG PRJEB41576 | [20] |
| Ruminal epithelial cells | Cell | RNA | GEO GSE166473 | [21] |
| Satellite cells | Cell | RNA | GEO GSE184128 | [22] |
| Satellite cells | Cell | RNA | GEO GSE211428 | [23] |
| Tissue(s) or Cell Type(s) | Isolation Type | Data Type | Database and Accession * | Reference |
|---|---|---|---|---|
| Conceptus | Cell | RNA | NCBI PRJNA987334 | [24] |
| Embryo | Cell | RNA | GEO GSE185233 | [25] |
| Ovary | Cell | RNA | GEO GSE233801 | [26] |
| Rumen | Cell | RNA | GSA CRA007511 | [27] |
| Rumen | Cell | RNA | NCBI PRJNA919098 | [28] |
| Skin | Cell | RNA | GEO GSE186204 | [29] |
| Testes | Cell | RNA | GEO GSE184343 | [30] |
| Testes | Cell | RNA | GSA CRA005236 | [31] |
| Tissue(s) or Cell Type(s) | Isolation Type | Data Type | Database and Accession * | Reference |
|---|---|---|---|---|
| Fetal Gonad | Cell | RNA, ATAC | GSA CRA006304, CRA006365 | [32] |
| Lung | Nuclei | RNA | GEO GSE183300 | [33] |
| Ovarian Follicle | Cell | RNA | GEO GSE135688 | [34] |
| Ovary | Cell | RNA | NCBI PRJNA1010653 | [35] |
| Rumen | Cell | RNA | NCBI PRJNA919098 | [28] |
| Skin | Cell | RNA | GEO GSE144351 | [36] |
| Skin | Cell | RNA | GEO GSE182474 | [37] |
| Skin | Cell | RNA | GEO GSE141284 | [38] |
| Testes | Cell | RNA | GEO GSE234407 | [39] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyons, A.; Brown, J.; Davenport, K.M. Single-Cell Sequencing Technology in Ruminant Livestock: Challenges and Opportunities. Curr. Issues Mol. Biol. 2024, 46, 5291-5306. https://doi.org/10.3390/cimb46060316
Lyons A, Brown J, Davenport KM. Single-Cell Sequencing Technology in Ruminant Livestock: Challenges and Opportunities. Current Issues in Molecular Biology. 2024; 46(6):5291-5306. https://doi.org/10.3390/cimb46060316
Chicago/Turabian StyleLyons, Avery, Jocelynn Brown, and Kimberly M. Davenport. 2024. "Single-Cell Sequencing Technology in Ruminant Livestock: Challenges and Opportunities" Current Issues in Molecular Biology 46, no. 6: 5291-5306. https://doi.org/10.3390/cimb46060316
APA StyleLyons, A., Brown, J., & Davenport, K. M. (2024). Single-Cell Sequencing Technology in Ruminant Livestock: Challenges and Opportunities. Current Issues in Molecular Biology, 46(6), 5291-5306. https://doi.org/10.3390/cimb46060316