
Targeting Cancers with oHSV-Based Oncolytic Viral Immunotherapy
Abstract
1. Introduction
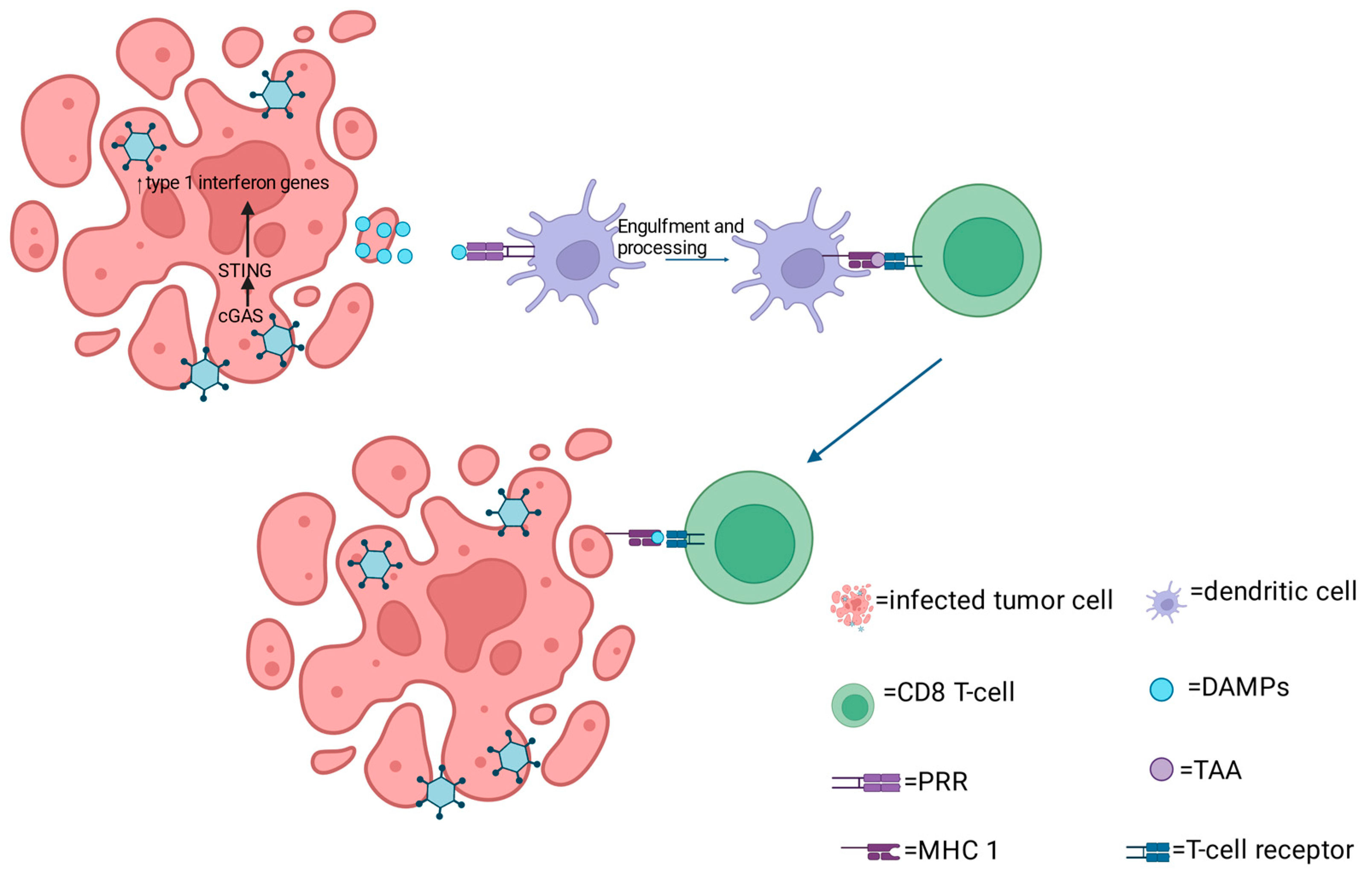
2. Mechanism of oHSV Anti-Tumor Activity
3. oHSV in Preclinical Models and Clinical Trials (Table 1)
3.1. Talimogene Laherparevec (T-VEC or Imlygic)
3.2. Delytact (Teserpaturev or G47∆)
3.3. HSV1716 (Seprehvir)

{kind=link}
{kind=link}
| Name | Strain and Mutation | Mechanism | Clinical Trials | Patient Group | Major Findings | Reference |
|---|---|---|---|---|---|---|
| T-VEC | HSV-1 JS-1 strain, which is modified to contain deletions of both infected cell protein 34.55 (also known as neurovirulence factor ICP34.5) and ICP47 genes. | Enhances antigen presentation through the TAP machinery. The virus expresses GM-CSF, which functions to promote the recruitment of APCs. | Phase I | 30 patients with Stage IIIC or IV melanoma | The virus was safe and well tolerated following intratumoral injection in patients with melanoma. | [36] |
| Phase II NCT00289016 | 50 patients with stage IIIC to IV melanoma | Intralesional administration of the virus resulted to an ORR of 26%, with responses observed in both injected and uninjected lesion, including visceral lesions. | [37] | |||
| Phase III NCT00769704a | 436 patients with unresected stage IIIB to IV melanoma | T-VEC provided a significant OS benefit to treated patients compared to GM-CSF therapy. | [12] | |||
| Phase III NCT02263508 | 692 patients with unresectable stage IIIB/IVM1c melanoma | No significant difference in ORR between T-VEC + pembrolizumab vs. placebo + pembrolizumab. No significant difference in safety profile of T-VEC + pembrolizumab vs. placebo + pembrolizumab. | [40] | |||
| G47∆ | HSV-1 F strain, which contains deletion of the gene encoding ICP47, to the genome of parental G207 virus, which contains deletions in both copies of the gene encoding for ICP34.5 and an inactivation of the gene encoding for ICP6. | The gene which encodes ICP6 which isa subunit of ribonucleotide reductase, an enzyme important for nucleotide metabolism and viral DNA replication in quiescent cells [42]. The inactivation of the gene encoding for ICP6 further restricts the virus to selectively replicate in tumor cells. | Phase I UMIN000002661 | 13 patients with Recurrent/progressive GBM | G47Δ was safe when inoculated into the brain of a human. | [52] |
| Phase II UMIN00001 5995 | 19 patients with residual or recurrent GBM | The virus was well tolerated, and the main side effects reported were fever, nausea, vomiting, and lymphopenia. Biopsy histology confirmed the presence of a larger number of tumor-infiltrating CD4+ and CD8+ T-cells and persistent low numbers of FoxP3+ cells following repeated G47∆ treatments. | [53] | |||
| Phase I UMIN000010463 | castration-resistant prostate cancer | G47∆ safety has been examined in other cancers. | [52] | |||
| Phase I jRCTs033180325 | olfactory neuroblastoma | G47∆ safety has been examined in other cancers. | [52] | |||
| Phase I jRCTs033180326 | malignant pleural mesothelioma | G47∆ safety has been examined in other cancers. | [52] | |||
| HSV1716 | HSV-1 strain 17, whichpossesses a deletion in the gene encoding the neurovirulence factor ICP34.5 | HSV1716 OV promotes the recruitment of effector immune cells into the TME through the upregulation of chemokines: monokines induced by IFN-γ (MIG also known as CXCL9) and IFN-γ inducible protein-10 (IP-10 also known as CXCL10). | Pilot study | five patients with stage IV metastatic melanoma | Immunohistochemical staining of injected nodules showed that viral replication was restrained to tumor cells. | [59] |
| Phase I | Nine patients with GBM | No adverse clinical symptoms or latent viral reactivation due to HSV1716 administration. | [60] |
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Kroschinsky, F.; Stolzel, F.; von Bonin, S.; Beutel, G.; Kochanek, M.; Kiehl, M.; Schellongowski, P. Intensive Care in Hematological and Oncological Patients (iCHOP) Collaborative Group. New drugs, new toxicities: Severe side effects of modern targeted and immunotherapy of cancer and their management. Crit. Care 2017, 21, 89. [Google Scholar] [CrossRef]
- de Vries, C.R.; Kaufman, H.L.; Lattime, E.C. Oncolytic viruses: Focusing on the tumor microenvironment. Cancer Gene Ther. 2015, 22, 169–171. [Google Scholar] [CrossRef]
- Koske, I.; Rossler, A.; Pipperger, L.; Petersson, M.; Barnstorf, I.; Kimpel, J.; Tripp, C.H.; Stoitzner, P.; Banki, Z.; von Laer, D. Oncolytic virotherapy enhances the efficacy of a cancer vaccine by modulating the tumor microenvironment. Int. J. Cancer 2019, 145, 1958–1969. [Google Scholar] [CrossRef]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef]
- Uche, I.K.; Kousoulas, K.G.; Rider, P.J.F. The Effect of Herpes Simplex Virus-Type-1 (HSV-1) Oncolytic Immunotherapy on the Tumor Microenvironment. Viruses 2021, 13, 1200. [Google Scholar] [CrossRef]
- Sugawara, K.; Iwai, M.; Ito, H.; Tanaka, M.; Seto, Y.; Todo, T. Oncolytic herpes virus G47Delta works synergistically with CTLA-4 inhibition via dynamic intratumoral immune modulation. Mol. Ther. Oncolytics 2021, 22, 129–142. [Google Scholar] [CrossRef]
- Wei, D.; Xu, J.; Liu, X.Y.; Chen, Z.N.; Bian, H. Fighting Cancer with Viruses: Oncolytic Virus Therapy in China. Hum. Gene Ther. 2018, 29, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Rahman, M.M.; McFadden, G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers 2021, 13, 5452. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.J.F.; Uche, I.K.; Sweeny, L.; Kousoulas, K.G. Anti-viral immunity in the tumor microenvironment: Implications for the rational design of herpes simplex virus type 1 oncolytic virotherapy. Curr. Clin. Microbiol. Rep. 2019, 6, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Senior, M. Checkpoint inhibitors go viral. Nat. Biotechnol. 2019, 37, 12–17. [Google Scholar] [CrossRef]
- Uche, I.K.; Fowlkes, N.; Vu, L.; Watanabe, T.; Carossino, M.; Nabi, R.; Del Piero, F.; Rudd, J.S.; Kousoulas, K.G.; Rider, P.J.F. Novel Oncolytic Herpes Simplex Virus 1 VC2 Promotes Long-Lasting, Systemic Anti-melanoma Tumor Immune Responses and Increased Survival in an Immunocompetent B16F10-Derived Mouse Melanoma Model. J. Virol. 2021, 95, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Markert, J.M.; Leavenworth, J.W. Modulation of the Intratumoral Immune Landscape by Oncolytic Herpes Simplex Virus Virotherapy. Front. Oncol. 2017, 7, 136. [Google Scholar] [CrossRef]
- Takasu, A.; Masui, A.; Hamada, M.; Imai, T.; Iwai, S.; Yura, Y. Immunogenic cell death by oncolytic herpes simplex virus type 1 in squamous cell carcinoma cells. Cancer Gene Ther. 2016, 23, 107–113. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Zloza, A.; Rabkin, S.D.; Kaufman, H.L. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology 2019, 8, 1591875. [Google Scholar] [CrossRef]
- Lee, J.; Ghonime, M.G.; Wang, R.; Cassady, K.A. The Antiviral Apparatus: STING and Oncolytic Virus Restriction. Mol. Ther. Oncolytics 2019, 13, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Froechlich, G.; Caiazza, C.; Gentile, C.; D’Alise, A.M.; De Lucia, M.; Langone, F.; Leoni, G.; Cotugno, G.; Scisciola, V.; Nicosia, A.; et al. Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus. Cancers 2020, 12, 3407. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pourchet, A.; Fuhrmann, S.R.; Pilones, K.A.; Demaria, S.; Frey, A.B.; Mulvey, M.; Mohr, I. CD8(+) T-cell Immune Evasion Enables Oncolytic Virus Immunotherapy. EBioMedicine 2016, 5, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Uche, I.K.; Stanfield, B.A.; Rudd, J.S.; Kousoulas, K.G.; Rider, P.J.F. Utility of a Recombinant HSV-1 Vaccine Vector for Personalized Cancer Vaccines. Front. Mol. Biosci. 2022, 9, 832393. [Google Scholar] [CrossRef]
- Nabi, R.; Musarrat, F.; Menk, P.L.J.C.; Langohr, I.M.; Chouljenko, V.N.; Kousoulas, K.G. The Oncolytic herpes simplex virus type-1 (HSV-1) vaccine strain VC2 causes intratumor infiltration of functionally active T cells and inhibition of tumor metastasis and pro-tumor genes VEGF and PDL1 expression in the 4T1/Balb/c mouse model of stage four breast cancer. Front. Mol. Biosci. 2023, 10, 1199068. [Google Scholar]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gogenur, I. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Wang, P.Y.; Hutzen, B.; Sprague, L.; Swain, H.M.; Love, J.K.; Stanek, J.R.; Boon, L.; Conner, J.; Cripe, T.P. Cooperation of Oncolytic Herpes Virotherapy and PD-1 Blockade in Murine Rhabdomyosarcoma Models. Sci. Rep. 2017, 7, 2396. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, K.; Chen, W.; Johnson, D.C.; Hendricks, R.L. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J. Exp. Med. 1998, 187, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Moesta, A.K.; Cooke, K.; Piasecki, J.; Mitchell, P.; Rottman, J.B.; Fitzgerald, K.; Zhan, J.; Yang, B.; Le, T.; Belmontes, B.; et al. Local Delivery of OncoVEX(mGM-CSF) Generates Systemic Antitumor Immune Responses Enhanced by Cytotoxic T-Lymphocyte-Associated Protein Blockade. Clin. Cancer Res. 2017, 23, 6190–6202. [Google Scholar] [CrossRef]
- Hu, J.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.A.; Ribas, A.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; Rutkowski, P.; et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined With Pembrolizumab for Advanced Melanoma. J. Clin. Oncol. 2023, 41, 528–540. [Google Scholar] [CrossRef]
- Scanlan, H.; Coffman, Z.; Bettencourt, J.; Shipley, T.; Bramblett, D.E. Herpes simplex virus 1 as an oncolytic viral therapy for refractory cancers. Front. Oncol. 2022, 12, 940019. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Shalhout, S.Z.; Iodice, G. Talimogene Laherparepvec: Moving From First-In-Class to Best-In-Class. Front. Mol. Biosci. 2022, 9, 834841. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.J.; Weller, S.K. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: Isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 1988, 62, 196–205. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Martuza, R.L.; Rabkin, S.D.; Ito, Y.; Todo, T. Oncolytic herpes simplex virus vector g47delta in combination with androgen ablation for the treatment of human prostate adenocarcinoma. Clin. Cancer Res. 2005, 11, 7886–7890. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.N.; Hu, P.; Zeng, M.S.; Liu, R.B. Anti-tumor effect of oncolytic herpes simplex virus G47delta on human nasopharyngeal carcinoma. Chin. J. Cancer 2011, 30, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Martuza, R.L.; Rabkin, S.D. Intracarotid delivery of oncolytic HSV vector G47Delta to metastatic breast cancer in the brain. Gene Ther. 2005, 12, 647–654. [Google Scholar] [CrossRef]
- Yajima, S.; Sugawara, K.; Iwai, M.; Tanaka, M.; Seto, Y.; Todo, T. Efficacy and safety of a third-generation oncolytic herpes virus G47Delta in models of human esophageal carcinoma. Mol. Ther. Oncolytics 2021, 23, 402–411. [Google Scholar] [CrossRef]
- Wang, J.; Xu, L.; Zeng, W.; Hu, P.; Zeng, M.; Rabkin, S.D.; Liu, R. Treatment of human hepatocellular carcinoma by the oncolytic herpes simplex virus G47delta. Cancer Cell Int. 2014, 14, 83. [Google Scholar] [CrossRef]
- Prabhakar, S.; Messerli, S.M.; Stemmer-Rachamimov, A.O.; Liu, T.C.; Rabkin, S.; Martuza, R.; Breakefield, X.O. Treatment of implantable NF2 schwannoma tumor models with oncolytic herpes simplex virus G47Delta. Cancer Gene Ther. 2007, 14, 460–467. [Google Scholar] [CrossRef]
- Antoszczyk, S.; Spyra, M.; Mautner, V.F.; Kurtz, A.; Stemmer-Rachamimov, A.O.; Martuza, R.L.; Rabkin, S.D. Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro Oncol. 2014, 16, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ino, Y.; Ohtsu, H.; Shibahara, J.; Tanaka, M. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat. Commun. 2022, 13, 4119. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Sever, O.N.; Oktay, K.; Guzel, E.; Kaya, V.; Guzel, A.; Yildirim, M. Reoperation does not provide a survival advantage in patients with recurrent Glioblastoma treated with irinotecan/bevacizumab treatment. Indian. J. Cancer 2021, 58, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Fraser, N.W. Role of the immune response during neuro-attenuated herpes simplex virus-mediated tumor destruction in a murine intracranial melanoma model. Cancer Res. 2000, 60, 5714–5722. [Google Scholar] [PubMed]
- Thomas, D.L.; Fraser, N.W. HSV-1 therapy of primary tumors reduces the number of metastases in an immune-competent model of metastatic breast cancer. Mol. Ther. 2003, 8, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courreges, M.C.; Conejo-Garcia, J.R.; Buckanovich, R.J.; Zhang, L.; Carroll, R.H.; Morgan, M.A.; Coukos, G. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum. Gene Ther. 2005, 16, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.; Winder, N.; Atkinson, E.; Al-Janabi, H.; Allen, R.J.; Hughes, R.; Moamin, M.; Louie, R.; Evans, D.; Hutchinson, M.; et al. Macrophages Mediate the Antitumor Effects of the Oncolytic Virus HSV1716 in Mammary Tumors. Mol. Cancer Ther. 2020, 20, 589–601. [Google Scholar] [CrossRef] [PubMed]
- MacKie, R.M.; Stewart, B.; Brown, S.M. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet 2001, 357, 525–526. [Google Scholar] [CrossRef]
- Rampling, R.; Cruickshank, G.; Papanastassiou, V.; Nicoll, J.; Hadley, D.; Brennan, D.; Petty, R.; MacLean, A.; Harland, J.; McKie, E.; et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000, 7, 859–866. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasar, R.T.; Uche, I.K.; Kousoulas, K.G. Targeting Cancers with oHSV-Based Oncolytic Viral Immunotherapy. Curr. Issues Mol. Biol. 2024, 46, 5582-5594. https://doi.org/10.3390/cimb46060334
Nasar RT, Uche IK, Kousoulas KG. Targeting Cancers with oHSV-Based Oncolytic Viral Immunotherapy. Current Issues in Molecular Biology. 2024; 46(6):5582-5594. https://doi.org/10.3390/cimb46060334
Chicago/Turabian StyleNasar, Rakin Tammam, Ifeanyi Kingsley Uche, and Konstantin G. Kousoulas. 2024. "Targeting Cancers with oHSV-Based Oncolytic Viral Immunotherapy" Current Issues in Molecular Biology 46, no. 6: 5582-5594. https://doi.org/10.3390/cimb46060334
APA StyleNasar, R. T., Uche, I. K., & Kousoulas, K. G. (2024). Targeting Cancers with oHSV-Based Oncolytic Viral Immunotherapy. Current Issues in Molecular Biology, 46(6), 5582-5594. https://doi.org/10.3390/cimb46060334