
The Role of Alarmins in the Pathogenesis of Atherosclerosis and Myocardial Infarction
 , ,
, ,
Abstract
:1. Introduction
2. Alarmins and Atherosclerosis
2.1. HMGB1
2.2. S100 Proteins
2.3. Interleukin-33
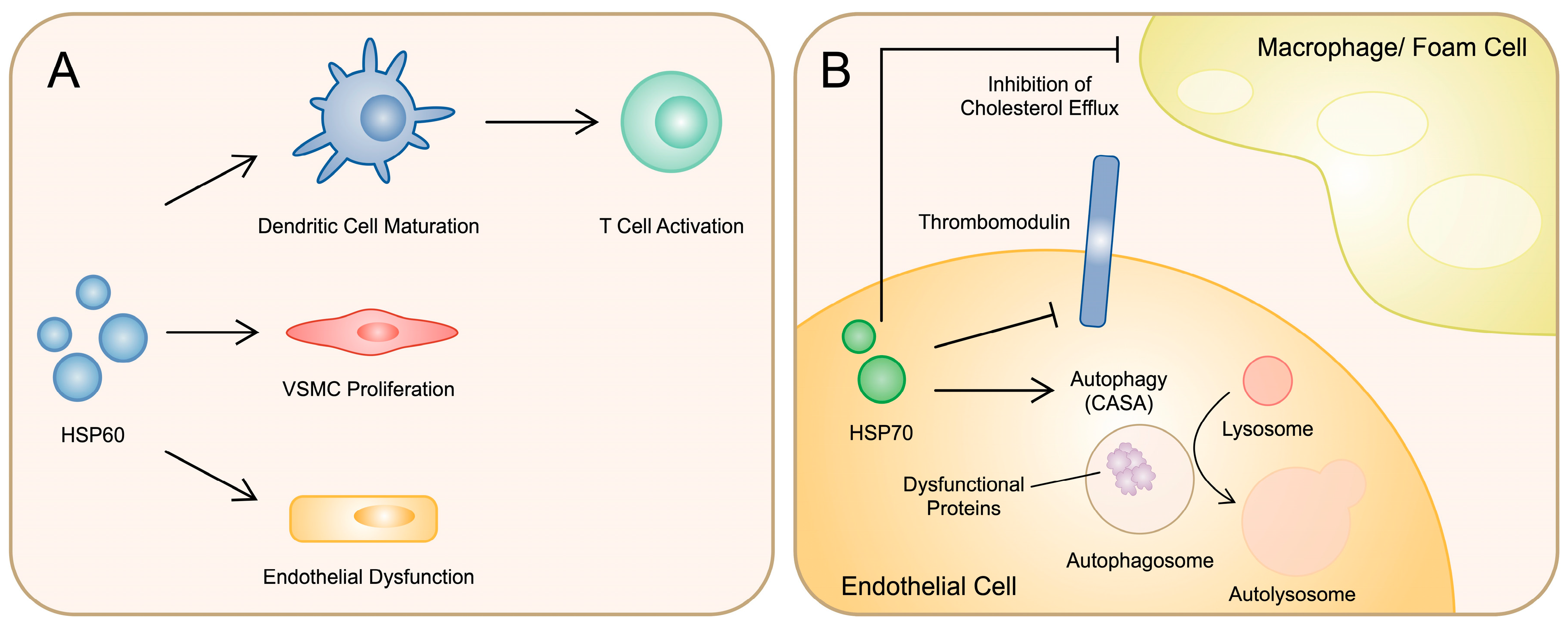
2.4. Heat Shock Proteins
3. Alarmins and Myocardial Infarction
3.1. HMGB1
3.2. Heat Shock Proteins
3.3. S100 Proteins
3.4. Interleukin-33
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Fang, Z.; Wang, H.; Cai, Y.; Rahimi, K.; Zhu, Y.; Fowkes, F.G.R.; Fowkes, F.J.I.; Rudan, I. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: A systematic review, meta-analysis, and modelling study. Lancet Glob. Health 2020, 8, e721–e729. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Orecchioni, M.; Ley, K. How the immune system shapes atherosclerosis: Roles of innate and adaptive immunity. Nat. Rev. Immunol. 2022, 22, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; McVey, D.G.; Shen, D.; Huang, X.; Ye, S. Phenotypic Switching of Vascular Smooth Muscle Cells in Atherosclerosis. J. Am. Heart Assoc. 2023, 12, e031121. [Google Scholar] [CrossRef]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Han, Z.; Oppenheim, J.J. Alarmins and immunity. Immunol. Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Viemann, D. S100-Alarmins Are Essential Pilots of Postnatal Innate Immune Adaptation. Front. Immunol. 2020, 11, 688. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.M.; Subbannayya, Y.; Rex, D.A.B.; Raju, R.; Chatterjee, O.; Advani, J.; Radhakrishnan, A.; Keshava Prasad, T.S.; Wani, M.R.; Pandey, A. A network map of IL-33 signaling pathway. J. Cell Commun. Signal 2018, 12, 615–624. [Google Scholar] [CrossRef]
- Kiełbowski, K.; Stańska, W.; Bakinowska, E.; Rusiński, M.; Pawlik, A. The Role of Alarmins in the Pathogenesis of Rheumatoid Arthritis, Osteoarthritis, and Psoriasis. Curr. Issues Mol. Biol. 2024, 46, 3640–3675. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Wu, Y.; Zhao, L.; Guo, J.; Zhang, K.; Chen, Z. Atorvastatin reduces serum HMGB1 levels in patients with hyperlipidemia. Exp. Ther. Med. 2012, 4, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.X.; Lu, L.; Peng, W.H.; Wang, L.J.; Zhang, Q.; Zhang, R.Y.; Chen, Q.J.; Shen, W.F. Increased serum HMGB1 level is associated with coronary artery disease in nondiabetic and type 2 diabetic patients. Atherosclerosis 2009, 205, 544–548. [Google Scholar] [CrossRef]
- Andrassy, M.; Volz, H.C.; Maack, B.; Schuessler, A.; Gitsioudis, G.; Hofmann, N.; Laohachewin, D.; Wienbrandt, A.R.; Kaya, Z.; Bierhaus, A.; et al. HMGB1 is associated with atherosclerotic plaque composition and burden in patients with stable coronary artery disease. PLoS ONE 2012, 7, e52081. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.W.; Zheng, X.X.; Zhou, T.; Tong, X.H.; Luo, C.Y.; Wang, X.A. HMGB1Modulates the Treg/Th17 Ratio in Atherosclerotic Patients. J. Atheroscler. Thromb. 2016, 23, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.L.; Wu, J.Y.; Tsai, C.S.; Lin, C.Y.; Tsai, Y.T.; Lin, C.S.; Wang, Y.F.; Yet, S.F.; Hsu, Y.J.; Kuo, C.C. TLR4-Activated MAPK-IL-6 Axis Regulates Vascular Smooth Muscle Cell Function. Int. J. Mol. Sci. 2016, 17, 1394. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, W.; Zhang, D. Gycyrrhizic acid alleviates atherosclerotic lesions in rats with diabetes mellitus. Mol. Med. Rep. 2021, 24, 755. [Google Scholar] [CrossRef]
- Inoue, K.; Kawahara, K.; Biswas, K.K.; Ando, K.; Mitsudo, K.; Nobuyoshi, M.; Maruyama, I. HMGB1 expression by activated vascular smooth muscle cells in advanced human atherosclerosis plaques. Cardiovasc. Pathol. 2007, 16, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Samah, N.; Ugusman, A.; Hamid, A.A.; Sulaiman, N.; Aminuddin, A. Role of Matrix Metalloproteinase-2 in the Development of Atherosclerosis among Patients with Coronary Artery Disease. Mediat. Inflamm. 2023, 2023, 9715114. [Google Scholar] [CrossRef]
- Eun, S.Y.; Ko, Y.S.; Park, S.W.; Chang, K.C.; Kim, H.J. IL-1β enhances vascular smooth muscle cell proliferation and migration via P2Y2 receptor-mediated RAGE expression and HMGB1 release. Vasc. Pharmacol. 2015, 72, 108–117. [Google Scholar] [CrossRef]
- Kim, E.J.; Park, S.Y.; Baek, S.E.; Jang, M.A.; Lee, W.S.; Bae, S.S.; Kim, K.; Kim, C.D. HMGB1 Increases IL-1β Production in Vascular Smooth Muscle Cells via NLRP3 Inflammasome. Front. Physiol. 2018, 9, 313. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Wu, D.; Sun, Y.; Suo, Y.; Yu, Q.; Zeng, M.; Gao, Q.; Yu, B.; Jiang, X.; Wang, Y. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci. Rep. 2021, 11, 19305. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wu, W.; Li, W.; Huang, S.; Li, Z.; Liu, R.; Shan, Z.; Zhang, C.; Wang, S. Activation of NLRP3 Inflammasome Promotes Foam Cell Formation in Vascular Smooth Muscle Cells and Atherogenesis Via HMGB1. J. Am. Heart Assoc. 2018, 7, e008596. [Google Scholar] [CrossRef]
- Hu, N.; Kong, L.; Qian, A.; Meng, Q.; Li, C.; Yu, X.; Chen, H.; Du, X.; Li, X. HMGB1 Silencing Potentiates the Anti-inflammatory Effects of Sodium Ferulate in ox-LDL-Stimulated Vascular Smooth Muscle Cells. Cell Biochem. Biophys. 2015, 72, 297–304. [Google Scholar] [CrossRef]
- Yeh, J.L.; Hsu, J.H.; Liang, J.C.; Chen, I.J.; Liou, S.F. Lercanidipine and labedipinedilol—A attenuate lipopolysaccharide/interferon-γ-induced inflammation in rat vascular smooth muscle cells through inhibition of HMGB1 release and MMP-2, 9 activities. Atherosclerosis 2013, 226, 364–372. [Google Scholar] [CrossRef]
- Zhang, T.; Li, H.; Ouyang, C.; Cao, G.; Gao, J.; Wu, J.; Yang, J.; Yu, N.; Min, Q.; Zhang, C.; et al. Liver kinase B1 inhibits smooth muscle calcification via high mobility group box 1. Redox Biol. 2021, 38, 101828. [Google Scholar] [CrossRef]
- Kiełbowski, K.; Bakinowska, E.; Procyk, G.; Ziętara, M.; Pawlik, A. The Role of MicroRNA in the Pathogenesis of Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2024, 25, 6108. [Google Scholar] [CrossRef]
- Guo, Y.; Luo, F.; Liu, Q.; Xu, D. Regulatory non-coding RNAs in acute myocardial infarction. J. Cell Mol. Med. 2017, 21, 1013–1023. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Chen, B.; Yang, F.; Hao, Z. miR-141-5p suppresses vascular smooth muscle cell inflammation, proliferation, and migration via inhibiting the HMGB1/NF-κB pathway. J. Biochem. Mol. Toxicol. 2021, 35, e22828. [Google Scholar] [CrossRef]
- Jiang, H.; Gong, R.; Wu, Y. miR-129-5p inhibits oxidized low-density lipoprotein-induced A7r5 cell viability and migration by targeting HMGB1 and the PI3k/Akt signaling pathway. Exp. Ther. Med. 2022, 23, 243. [Google Scholar] [CrossRef]
- Chen, L.B.; An, Z.; Zheng, H.K.; Wang, X.P.; Shan, R.T.; Mao, C.Y.; Zhang, W.Q. MicroRNA-34c suppresses proliferation of vascular smooth muscle cell via modulating high mobility group box protein 1. J. Clin. Lab. Anal. 2020, 34, e23293. [Google Scholar] [CrossRef]
- Yang, J.; Chen, L.; Ding, J.; Fan, Z.; Li, S.; Wu, H.; Zhang, J.; Yang, C.; Wang, H.; Zeng, P. MicroRNA-24 inhibits high glucose-induced vascular smooth muscle cell proliferation and migration by targeting HMGB1. Gene 2016, 586, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Liu, Z.; Qi, X. LncRNA BANCR induced vascular smooth muscle cell proliferation by downregulating miR-34c methylation in atherosclerosis. J. Thromb. Thrombolysis 2021, 51, 924–932. [Google Scholar] [CrossRef]
- Guo, X.; Liu, Y.; Zheng, X.; Han, Y.; Cheng, J. HOTTIP knockdown inhibits cell proliferation and migration via regulating miR-490-3p/HMGB1 axis and PI3K-AKT signaling pathway in ox-LDL-induced VSMCs. Life Sci. 2020, 248, 117445. [Google Scholar] [CrossRef]
- Ding, P.; Ding, Y.; Tian, Y.; Lei, X. Circular RNA circ_0010283 regulates the viability and migration of oxidized low-density lipoprotein-induced vascular smooth muscle cells via an miR-370-3p/HMGB1 axis in atherosclerosis. Int. J. Mol. Med. 2020, 46, 1399–1408. [Google Scholar] [CrossRef]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef]
- Chen, Z.; Pan, X.; Sheng, Z.; Yan, G.; Chen, L.; Ma, G. Baicalin Suppresses the Proliferation and Migration of Ox-LDL-VSMCs in Atherosclerosis through Upregulating miR-126-5p. Biol. Pharm. Bull. 2019, 42, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Harja, E.; Bu, D.X.; Hudson, B.I.; Chang, J.S.; Shen, X.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J. Clin. Investig. 2008, 118, 183–194. [Google Scholar] [CrossRef]
- Gao, H.; Li, H.; Li, W.; Shen, X.; Di, B. Pioglitazone Attenuates Atherosclerosis in Diabetic Mice by Inhibition of Receptor for Advanced Glycation End-Product (RAGE) Signaling. Med. Sci. Monit. 2017, 23, 6121–6131. [Google Scholar] [CrossRef]
- Bao, Z.; Li, L.; Geng, Y.; Yan, J.; Dai, Z.; Shao, C.; Sun, Z.; Jing, L.; Pang, Q.; Zhang, L.; et al. Advanced Glycation End Products Induce Vascular Smooth Muscle Cell-Derived Foam Cell Formation and Transdifferentiate to a Macrophage-Like State. Mediat. Inflamm. 2020, 2020, 6850187. [Google Scholar] [CrossRef]
- Chellan, B.; Reardon, C.A.; Getz, G.S.; Hofmann Bowman, M.A. Enzymatically Modified Low-Density Lipoprotein Promotes Foam Cell Formation in Smooth Muscle Cells via Macropinocytosis and Enhances Receptor-Mediated Uptake of Oxidized Low-Density Lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1101–1113. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef]
- Jeong, J.; Cho, S.; Seo, M.; Lee, B.S.; Jang, Y.; Lim, S.; Park, S. Soluble RAGE attenuates Ang II-induced arterial calcification via inhibiting AT1R-HMGB1-RAGE axis. Atherosclerosis 2022, 346, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P.; Camici, P.; et al. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010, 9, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Su, B.; Qin, G.; Zhao, L. HMGB1 promotes Ox-LDL-induced endothelial cell damage by inhibiting PI3K/Akt signaling pathway. BMC Cardiovasc. Disord. 2022, 22, 555. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, S.; Jang, E.; Naderinabi, F.; Sanwal, R.; Khosraviani, N.; Wang, C.; Steinberg, B.E.; Goldenberg, N.M.; Ikeda, J.; Lee, W.L. Endothelial HMGB1 Is a Critical Regulator of LDL Transcytosis via an SREBP2-SR-BI Axis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 200–216. [Google Scholar] [CrossRef]
- Hou, C.; Jiang, X.; Sheng, W.; Zhang, Y.; Lin, Q.; Hong, S.; Zhao, J.; Wang, T.; Ye, X. Xinmaikang (XMK) tablets alleviate atherosclerosis by regulating the SREBP2-mediated NLRP3/ASC/Caspase-1 signaling pathway. J. Ethnopharmacol. 2024, 319 Pt 2, 117240. [Google Scholar] [CrossRef]
- Bai, X.; Wang, S.; Shu, L.; Cao, Q.; Hu, H.; Zhu, Y.; Chen, C. Hawthorn leaf flavonoids alleviate the deterioration of atherosclerosis by inhibiting SCAP-SREBP2-LDLR pathway through sPLA2-IIA signaling in macrophages in mice. J. Ethnopharmacol. 2024, 327, 118006. [Google Scholar] [CrossRef]
- Tan, W.; Wang, G.; Liu, G.; You, D.; Wei, M.; Jin, X.; Zhao, W.; Zheng, M. The elevation of miR-185-5p alleviates high-fat diet-induced atherosclerosis and lipid accumulation. Aging 2022, 14, 1729–1742. [Google Scholar] [CrossRef]
- Kim, S.; Woo, C.H. Laminar Flow Inhibits ER Stress-Induced Endothelial Apoptosis through PI3K/Akt-Dependent Signaling Pathway. Mol. Cells 2018, 41, 964–970. [Google Scholar] [CrossRef]
- Luo, Y.; Li, S.J.; Yang, J.; Qiu, Y.Z.; Chen, F.P. HMGB1 induces an inflammatory response in endothelial cells via the RAGE-dependent endoplasmic reticulum stress pathway. Biochem. Biophys. Res. Commun. 2013, 438, 732–738. [Google Scholar] [CrossRef]
- Savransky, V.; Nanayakkara, A.; Li, J.; Bevans, S.; Smith, P.L.; Rodriguez, A.; Polotsky, V.Y. Chronic intermittent hypoxia induces atherosclerosis. Am. J. Respir. Crit. Care Med. 2007, 175, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Lee, J.; Lim, J.; Cho, S.; An, S.; Lee, M.; Yoon, N.; Seo, M.; Lim, S.; Park, S. Soluble RAGE attenuates AngII-induced endothelial hyperpermeability by disrupting HMGB1-mediated crosstalk between AT1R and RAGE. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.H.; Jian, W.X.; Li, H.L.; Hou, L.; Wei, Y.D.; Li, W.M.; Xu, Y.W. Increased serum myeloid-related protein 8/14 level is associated with atherosclerosis in type 2 diabetic patients. Cardiovasc. Diabetol. 2011, 10, 41. [Google Scholar] [CrossRef]
- Cotoi, O.S.; Dunér, P.; Ko, N.; Hedblad, B.; Nilsson, J.; Björkbacka, H.; Schiopu, A. Plasma S100A8/A9 correlates with blood neutrophil counts, traditional risk factors, and cardiovascular disease in middle-aged healthy individuals. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O. Multiple roles for neutrophils in atherosclerosis. Circ. Res. 2012, 110, 875–888. [Google Scholar] [CrossRef]
- Miyamoto, S.; Ueda, M.; Ikemoto, M.; Naruko, T.; Itoh, A.; Tamaki, S.; Nohara, R.; Terasaki, F.; Sasayama, S.; Fujita, M. Increased serum levels and expression of S100A8/A9 complex in infiltrated neutrophils in atherosclerotic plaque of unstable angina. Heart 2008, 94, 1002–1007. [Google Scholar] [CrossRef]
- Natorska, J.; Ząbczyk, M.; Undas, A. Neutrophil extracellular traps (NETs) in cardiovascular diseases: From molecular mechanisms to therapeutic interventions. Kardiol. Pol. 2023, 81, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Sprenkeler, E.G.G.; Zandstra, J.; van Kleef, N.D.; Goetschalckx, I.; Verstegen, B.; Aarts, C.E.M.; Janssen, H.; Tool, A.T.J.; van Mierlo, G.; van Bruggen, R.; et al. S100A8/A9 Is a Marker for the Release of Neutrophil Extracellular Traps and Induces Neutrophil Activation. Cells 2022, 11, 236. [Google Scholar] [CrossRef]
- Hofmann Bowman, M.; Wilk, J.; Heydemann, A.; Kim, G.; Rehman, J.; Lodato, J.A.; Raman, J.; McNally, E.M. S100A12 mediates aortic wall remodeling and aortic aneurysm. Circ. Res. 2010, 106, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Hofmann Bowman, M.A.; Gawdzik, J.; Bukhari, U.; Husain, A.N.; Toth, P.T.; Kim, G.; Earley, J.; McNally, E.M. S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein E-null mice by activating an osteogenic gene regulatory program. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 337–344. [Google Scholar] [CrossRef]
- Chellan, B.; Yan, L.; Sontag, T.J.; Reardon, C.A.; Hofmann Bowman, M.A. IL-22 is induced by S100/calgranulin and impairs cholesterol efflux in macrophages by downregulating ABCG1. J. Lipid Res. 2014, 55, 443–454. [Google Scholar] [CrossRef]
- Li, L.; Mou, J.; Han, Y.; Wang, M.; Lu, S.; Ma, Q.; Wang, J.; Ye, J.; Sun, G. Calenduloside e modulates macrophage polarization via KLF2-regulated glycolysis, contributing to attenuates atherosclerosis. Int. Immunopharmacol. 2023, 117, 109730. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, S.A.; Neto, N.G.B.; Devilly, E.; Shanley, L.C.; Fitzgerald, H.K.; Monaghan, M.G.; Dunne, A. Cholesterol crystals drive metabolic reprogramming and M1 macrophage polarisation in primary human macrophages. Atherosclerosis 2022, 352, 35–45. [Google Scholar] [CrossRef]
- Stankovic, M.; Ljujic, B.; Babic, S.; Maravic-Stojkovic, V.; Mitrovic, S.; Arsenijevic, N.; Radak, D.; Pejnovic, N.; Lukic, M.L. IL-33/IL-33R in various types of carotid artery atherosclerotic lesions. Cytokine 2019, 120, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.M.; Xu, D.; Asquith, D.L.; Denby, L.; Li, Y.; Sattar, N.; Baker, A.H.; McInnes, I.B.; Liew, F.Y. IL-33 reduces the development of atherosclerosis. J. Exp. Med. 2008, 205, 339–346. [Google Scholar] [CrossRef]
- Gu, C.; Fan, X.; Yu, W. Functional Diversity of Mammalian Small Heat Shock Proteins: A Review. Cells 2023, 12, 1947. [Google Scholar] [CrossRef]
- Milani, A.; Basirnejad, M.; Bolhassani, A. Heat-shock proteins in diagnosis and treatment: An overview of different biochemical and immunological functions. Immunotherapy 2019, 11, 215–239. [Google Scholar] [CrossRef]
- Liao, B.H.; Xu, Z.L.; Gao, F.; Zhang, S.H.; Liang, R.J.; Dong, S.H. The relationship between HSP60 gene polymorphisms and susceptibility to atherosclerosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2667–2673. [Google Scholar] [CrossRef] [PubMed]
- Zonnar, S.; Saeedy, S.A.G.; Nemati, F.; Motamedi, M.J.; Raeespour, H.; Amani, J. Decrescent role of recombinant HSP60 antibody against atherosclerosis in high-cholesterol diet immunized rabbits. Iran. J. Basic. Med. Sci. 2022, 25, 32–38. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, Z.; Jiang, L.; Chen, F.; Jin, R.; Cheng, L. Effects of oral and subcutaneous administration of HSP60 on myeloid-derived suppressor cells and atherosclerosis in ApoE-/- mice. Biochem. Biophys. Res. Commun. 2018, 498, 701–706. [Google Scholar] [CrossRef]
- Li, Q.; Mei, A.; Qian, H.; Min, X.; Yang, H.; Zhong, J.; Li, C.; Xu, H.; Chen, J. The role of myeloid-derived immunosuppressive cells in cardiovascular disease. Int. Immunopharmacol. 2023, 117, 109955. [Google Scholar] [CrossRef] [PubMed]
- Mundkur, L.; Mukhopadhyay, R.; Samson, S.; Varma, M.; Kale, D.; Chen, D.; Shivaprasad, S.; Sivanandan, H.; Soman, V.; Lu, X.; et al. Mucosal tolerance to a combination of ApoB and HSP60 peptides controls plaque progression and stabilizes vulnerable plaque in Apob(tm2Sgy)Ldlr(tm1Her)/J mice. PLoS ONE 2013, 8, e58364. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Zhou, D.; Zhang, J.; Guo, G.; Chen, Y. Pathogenic Autoimmunity in Atherosclerosis Evolves from HSP60-Reactive CD4+ T Cells. J. Cardiovasc. Transl. Res. 2024; ahead of print. [Google Scholar] [CrossRef]
- Shirsath, K.; Joshi, A.; Vohra, A.; Devkar, R. HSP60 knockdown exerts differential response in endothelial cells and monocyte derived macrophages during atherogenic transformation. Sci. Rep. 2021, 11, 1086. [Google Scholar] [CrossRef]
- Martinus, R.D.; Goldsbury, J. Endothelial TNF-alpha induction by Hsp60 secreted from THP-1 monocytes exposed to hyperglycaemic conditions. Cell Stress Chaperones 2018, 23, 519–525. [Google Scholar] [CrossRef]
- Hinkley, H.; Counts, D.A.; VonCanon, E.; Lacy, M. T Cells in Atherosclerosis: Key Players in the Pathogenesis of Vascular Disease. Cells 2023, 12, 2152. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Steuer, J.; Gillgren, P.; Hayderi, A.; Liu, A.; Frostegard, J. Induction of Dendritic Cell-Mediated Activation of T Cells from Atherosclerotic Plaques by Human Heat Shock Protein 60. J. Am. Heart Assoc. 2017, 6, e006778. [Google Scholar] [CrossRef]
- Deniset, J.F.; Hedley, T.E.; Hlavackova, M.; Chahine, M.N.; Dibrov, E.; O’Hara, K.; Maddaford, G.G.; Nelson, D.; Maddaford, T.G.; Fandrich, R.; et al. Heat shock protein 60 involvement in vascular smooth muscle cell proliferation. Cell Signal 2018, 47, 44–51. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.J.; Li, Y.H.; Shi, G.Y.; Liu, S.L.; Chang, P.C.; Kuo, C.H.; Wu, H.L. Thrombomodulin domains attenuate atherosclerosis by inhibiting thrombin-induced endothelial cell activation. Cardiovasc. Res. 2011, 92, 317–327. [Google Scholar] [CrossRef]
- Araujo, T.L.S.; Venturini, G.; Moretti, A.I.S.; Tanaka, L.Y.; Pereira, A.C.; Laurindo, F.R.M. Cell-surface HSP70 associates with thrombomodulin in endothelial cells. Cell Stress Chaperones 2019, 24, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.W.; Zhang, M.; Chen, L.Y.; Gong, D.; Xia, X.D.; Yu, X.H.; Wang, S.Q.; Ou, X.; Dai, X.Y.; Zheng, X.L.; et al. Heat shock protein 70 accelerates atherosclerosis by downregulating the expression of ABCA1 and ABCG1 through the JNK/Elk-1 pathway. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 806–822. [Google Scholar] [CrossRef]
- Tedesco, B.; Vendredy, L.; Timmerman, V.; Poletti, A. The chaperone-assisted selective autophagy complex dynamics and dysfunctions. Autophagy 2023, 19, 1619–1641. [Google Scholar] [CrossRef]
- Diao, H.; Wu, K.; Lan, D.; Wang, D.; Zhao, J.; Huang, B.; Shao, X.; Wang, R.; Tan, H.; Tang, X.; et al. BAG3 Alleviates Atherosclerosis by Inhibiting Endothelial-to-Mesenchymal Transition via Autophagy Activation. Genes 2022, 13, 1338. [Google Scholar] [CrossRef]
- Qu, W.; Zhou, X.; Jiang, X.; Xie, X.; Xu, K.; Gu, X.; Na, R.; Piao, M.; Xi, X.; Sun, N.; et al. Long Noncoding RNA Gpr137b-ps Promotes Advanced Atherosclerosis via the Regulation of Autophagy in Macrophages. Arterioscler. Thromb. Vasc. Biol. 2023, 43, e468–e489. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Ding, X.; Meng, C.; Dong, H.; Zhang, S.; Zhou, H.; Tan, W.; Huang, L.; He, A.; Li, J.; Huang, J.; et al. Extracellular Hsp90alpha, which participates in vascular inflammation, is a novel serum predictor of atherosclerosis in type 2 diabetes. BMJ Open Diabetes Res. Care 2022, 10, e002579. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Tang, X.; Miao, Z.; Chen, Y.; Cao, J.; Song, T.; You, D.; Zhong, Y.; Lin, Z.; Wang, D.; et al. Hsp90 S-nitrosylation at Cys521, as a conformational switch, modulates cycling of Hsp90-AHA1-CDC37 chaperone machine to aggravate atherosclerosis. Redox Biol. 2022, 52, 102290. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Bullock, D.; Lim, A.; Argemi, J.; Orning, P.; Lien, E.; Bataller, R.; Mandrekar, P. Inhibition of HSP90 and Activation of HSF1 Diminish Macrophage NLRP3 Inflammasome Activity in Alcohol-Associated Liver Injury. Alcohol. Clin. Exp. Res. 2020, 44, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.A.; Akhter, M.S.; Kubra, K.T.; Barabutis, N. Hsp90 inhibition protects brain endothelial cells against LPS-induced injury. Biofactors 2022, 48, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Fu, S.; Zhan, X.; Wang, Z.; Zhang, P.; Shi, W.; Qin, N.; Chen, Y.; Wang, C.; Niu, M.; et al. Echinatin effectively protects against NLRP3 inflammasome-driven diseases by targeting HSP90. JCI Insight 2021, 6, e134601. [Google Scholar] [CrossRef] [PubMed]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D.; Mickley, H.; Crea, F.; Van de Werf, F.; et al. Fourth universal definition of myocardial infarction (2018). Kardiol. Pol. 2018, 76, 1383–1415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, W.; Liu, H. The Role of Immune Cells in Cardiac Remodeling after Myocardial Infarction. J. Cardiovasc. Pharmacol. 2020, 76, 407–413. [Google Scholar] [CrossRef]
- Saleh, M.; Ambrose, J.A. Understanding myocardial infarction. F1000Research 2018, 7, 1378. [Google Scholar] [CrossRef]
- Salari, N.; Morddarvanjoghi, F.; Abdolmaleki, A.; Rasoulpoor, S.; Khaleghi, A.A.; Hezarkhani, L.A.; Shohaimi, S.; Mohammadi, M. The global prevalence of myocardial infarction: A systematic review and meta-analysis. BMC Cardiovasc. Disord. 2023, 23, 206. [Google Scholar] [CrossRef]
- Rohani, C.; Jafarpoor, H.; Mortazavi, Y.; Esbakian, B.; Gholinia, H. Mortality in patients with myocardial infarction and potential risk factors: A five-year data analysis. ARYA Atheroscler. 2022, 18, 1–8. [Google Scholar] [CrossRef]
- Kologrivova, I.; Shtatolkina, M.; Suslova, T.; Ryabov, V. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair after Myocardial Infarction. Front. Immunol. 2021, 12, 664457. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Frangogiannis, N.G. Immune cells in repair of the infarcted myocardium. Microcirculation 2017, 24, e12305. [Google Scholar] [CrossRef]
- Ma, Y.; Yabluchanskiy, A.; Lindsey, M.L. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair. 2013, 6, 11. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Pittet, M.J.; Swirski, F.K. Monocytes: Protagonists of infarct inflammation and repair after myocardial infarction. Circulation 2010, 121, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Silva, V.; Frantz, S.; Ramos, G.C. Lymphocytes at the Heart of Wound Healing. Adv. Exp. Med. Biol. 2017, 1003, 225–250. [Google Scholar] [CrossRef]
- Kyaw, T.; Loveland, P.; Kanellakis, P.; Cao, A.; Kallies, A.; Huang, A.L.; Peter, K.; Toh, B.H.; Bobik, A. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur. Heart J. 2021, 42, 938–947. [Google Scholar] [CrossRef]
- Ghigo, A.; Franco, I.; Morello, F.; Hirsch, E. Myocyte signalling in leucocyte recruitment to the heart. Cardiovasc. Res. 2014, 102, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Loukili, N.; Rosenblatt-Velin, N.; Li, J.; Clerc, S.; Pacher, P.; Feihl, F.; Waeber, B.; Liaudet, L. Peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infarcted myocardium in vivo. Cardiovasc. Res. 2011, 89, 586–594. [Google Scholar] [CrossRef]
- de Haan, J.J.; Smeets, M.B.; Pasterkamp, G.; Arslan, F. Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediat. Inflamm. 2013, 2013, 206039. [Google Scholar] [CrossRef]
- Oyama, J.; Blais, C.; Liu, X.; Pu, M.; Kobzik, L.; Kelly, R.A.; Bourcier, T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation 2004, 109, 784–789. [Google Scholar] [CrossRef]
- Andrassy, M.; Volz, H.C.; Igwe, J.C.; Funke, B.; Eichberger, S.N.; Kaya, Z.; Buss, S.; Autschbach, F.; Pleger, S.T.; Lukic, I.K.; et al. High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation 2008, 117, 3216–3226. [Google Scholar] [CrossRef] [PubMed]
- Foglio, E.; Pellegrini, L.; Russo, M.A.; Limana, F. HMGB1-Mediated Activation of the Inflammatory-Reparative Response Following Myocardial Infarction. Cells 2022, 11, 216. [Google Scholar] [CrossRef]
- Karuppagounder, V.; Giridharan, V.V.; Arumugam, S.; Sreedhar, R.; Palaniyandi, S.S.; Krishnamurthy, P.; Quevedo, J.; Watanabe, K.; Konishi, T.; Thandavarayan, R.A. Modulation of Macrophage Polarization and HMGB1-TLR2/TLR4 Cascade Plays a Crucial Role for Cardiac Remodeling in Senescence-Accelerated Prone Mice. PLoS ONE 2016, 11, e0152922. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, T.; Takeishi, Y.; Harada, M.; Niizeki, T.; Suzuki, S.; Sasaki, T.; Ishino, M.; Bilim, O.; Nakajima, O.; Kubota, I. High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc. Res. 2008, 80, 40–46. [Google Scholar] [CrossRef]
- Limana, F.; Germani, A.; Zacheo, A.; Kajstura, J.; Di Carlo, A.; Borsellino, G.; Leoni, O.; Palumbo, R.; Battistini, L.; Rastaldo, R.; et al. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ. Res. 2005, 97, e73–e83. [Google Scholar] [CrossRef]
- Kohno, T.; Anzai, T.; Naito, K.; Miyasho, T.; Okamoto, M.; Yokota, H.; Yamada, S.; Maekawa, Y.; Takahashi, T.; Yoshikawa, T.; et al. Role of high-mobility group box 1 protein in post-infarction healing process and left ventricular remodelling. Cardiovasc. Res. 2009, 81, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Andrassy, M.; Volz, H.C.; Riedle, N.; Gitsioudis, G.; Seidel, C.; Laohachewin, D.; Zankl, A.R.; Kaya, Z.; Bierhaus, A.; Giannitsis, E.; et al. HMGB1 as a predictor of infarct transmurality and functional recovery in patients with myocardial infarction. J. Intern. Med. 2011, 270, 245–253. [Google Scholar] [CrossRef]
- Takahashi, K.; Fukushima, S.; Yamahara, K.; Yashiro, K.; Shintani, Y.; Coppen, S.R.; Salem, H.K.; Brouilette, S.W.; Yacoub, M.H.; Suzuki, K. Modulated Inflammation by Injection of High-Mobility Group Box 1 Recovers Post-Infarction Chronically Failing Heart. Circulation 2008, 118, S106–S114. [Google Scholar] [CrossRef]
- Woo, Y.J.; Taylor, M.D.; Cohen, J.E.; Jayasankar, V.; Bish, L.T.; Burdick, J.; Pirolli, T.J.; Berry, M.F.; Hsu, V.; Grand, T. Ethyl pyruvate preserves cardiac function and attenuates oxidative injury after prolonged myocardial ischemia. J. Thorac. Cardiovasc. Surg. 2004, 127, 1262–1269. [Google Scholar] [CrossRef]
- Lin, L.; Kim, S.C.; Wang, Y.; Gupta, S.; Davis, B.; Simon, S.I.; Torre-Amione, G.; Knowlton, A.A. HSP60 in heart failure: Abnormal distribution and role in cardiac myocyte apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2238–H2247. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Tang, H.; Mitchell-Silbaugh, K.; Fang, X.; Han, Z.; Ouyang, K. Heat Shock Protein 60 in Cardiovascular Physiology and Diseases. Front. Mol. Biosci. 2020, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Guo, X.; Liu, X.M.; Liu, L.; Weng, Q.F.; Dong, S.J.; Knowlton, A.A.; Yuan, W.J.; Lin, L. Extracellular HSP60 induces inflammation through activating and up-regulating TLRs in cardiomyocytes. Cardiovasc. Res. 2013, 98, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Si, R.; Feng, Y.; Chen, H.H.; Zou, L.; Wang, E.; Zhang, M.; Warren, H.S.; Sosnovik, D.E.; Chao, W. Myocardial ischemia activates an injurious innate immune signaling via cardiac heat shock protein 60 and Toll-like receptor 4. J. Biol. Chem. 2011, 286, 31308–31319. [Google Scholar] [CrossRef] [PubMed]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef]
- Wu, J.; Chen, S.; Liu, Y.; Liu, Z.; Wang, D.; Cheng, Y. Therapeutic perspectives of heat shock proteins and their protein-protein interactions in myocardial infarction. Pharmacol. Res. 2020, 160, 105162. [Google Scholar] [CrossRef]
- Song, Y.J.; Zhong, C.B.; Wang, X.B. Heat shock protein 70: A promising therapeutic target for myocardial ischemia-reperfusion injury. J. Cell Physiol. 2019, 234, 1190–1207. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, V.; Suresh, S.C.; Thirunavukkarasu, M.; Mannu, J.; Foye, J.L.C.; Mathur, P.P.; Palesty, J.A.; Sanchez, J.A.; McFadden, D.W.; Maulik, N. Regulation of A-Kinase-Anchoring Protein 12 by Heat Shock Protein A12B to Prevent Ventricular Dysfunction Following Acute Myocardial Infarction in Diabetic Rats. J. Cardiovasc. Transl. Res. 2017, 10, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.W.; Zhang, G.H.; Zhang, Y.Y.; Li, W.D.; Wang, C.H.; Yin, C.Y.; Zhang, F.M. Lipopolysaccharide pretreatment protects against ischemia/reperfusion injury via increase of HSP70 and inhibition of NF-κB. Cell Stress Chaperones 2011, 16, 287–296. [Google Scholar] [CrossRef]
- Latchman, D. Heat Shock Proteins and Cardiac Protection. Cardiovascular Research 2001, 51, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Lubbers, N.L.; Polakowski, J.S.; Wegner, C.D.; Burke, S.E.; Diaz, G.J.; Daniell, K.M.; Cox, B.F. Oral Bimoclomol Elevates Heat Shock Protein 70 and Reduces Myocardial Infarct Size in Rats. European Journal of Pharmacology 2002, 435, 79–83. [Google Scholar] [CrossRef]
- Bluhm, W.F.; Martin, J.L.; Mestril, R.; Dillmann, W.H. Specific heat shock proteins protect microtubules during simulated ischemia in cardiac myocytes. Am. J. Physiol. 1998, 275, H2243–H2249. [Google Scholar] [CrossRef] [PubMed]
- Murashov, A.K.; Haq, I.U.; Hill, C.; Park, E.; Smith, M.; Wang, X.; Wang, X.; Goldberg, D.J.; Wolgemuth, D.J. Crosstalk between p38, Hsp25 and Akt in spinal motor neurons after sciatic nerve injury. Brain Res. Mol. Brain Res. 2001, 93, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.A.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094. [Google Scholar] [CrossRef]
- Chalise, U.; Becirovic-Agic, M.; Daseke, M.J.; Konfrst, S.R.; Rodriguez-Paar, J.R.; Feng, D.; Salomon, J.D.; Anderson, D.R.; Cook, L.M.; Lindsey, M.L. S100A9 is a functional effector of infarct wall thinning after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H145–H155. [Google Scholar] [CrossRef]
- Li, Y.; Chen, B.; Yang, X.; Zhang, C.; Jiao, Y.; Li, P.; Liu, Y.; Li, Z.; Qiao, B.; Bond Lau, W.; et al. S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocyte Death in Response to Ischemic/Reperfusion Injury. Circulation 2019, 140, 751–764. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Marinković, G.; Grauen Larsen, H.; Yndigegn, T.; Szabo, I.A.; Mares, R.G.; de Camp, L.; Weiland, M.; Tomas, L.; Goncalves, I.; Nilsson, J.; et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. Eur. Heart J. 2019, 40, 2713–2723. [Google Scholar] [CrossRef]
- Boteanu, R.M.; Suica, V.I.; Uyy, E.; Ivan, L.; Cerveanu-Hogas, A.; Mares, R.G.; Simionescu, M.; Schiopu, A.; Antohe, F. Short-Term Blockade of Pro-Inflammatory Alarmin S100A9 Favorably Modulates Left Ventricle Proteome and Related Signaling Pathways Involved in Post-Myocardial Infarction Recovery. Int. J. Mol. Sci. 2022, 23, 5289. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Duan, X.; Wang, B.; Zhan, Z. Targeting NPM1 Epigenetically Promotes Postinfarction Cardiac Repair by Reprogramming Reparative Macrophage Metabolism. Circulation 2024, 149, 1982–2001. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Zhu, R.; Hou, X.; Wu, F.; Feng, R. Integrated Analysis Reveals S100a8/a9 Regulates Autophagy and Apoptosis through the MAPK and PI3K-AKT Signaling Pathway in the Early Stage of Myocardial Infarction. Cells 2022, 11, 1911. [Google Scholar] [CrossRef] [PubMed]
- Korshunova, A.Y.; Blagonravov, M.L.; Neborak, E.V.; Syatkin, S.P.; Sklifasovskaya, A.P.; Semyatov, S.M.; Agostinelli, E. BCL2-regulated apoptotic process in myocardial ischemia-reperfusion injury (Review). Int. J. Mol. Med. 2021, 47, 23–36. [Google Scholar] [CrossRef]
- Marinković, G.; Koenis, D.S.; de Camp, L.; Jablonowski, R.; Graber, N.; de Waard, V.; de Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xiang, Z.; Zhang, J.; Zhai, Y.; Fan, Z.; Wang, H.; Wu, J.; Huang, Y.; Xiong, M.; Ma, C. miR-24 Alleviates MI/RI by Blocking the S100A8/TLR4/MyD88/NF-κB Pathway. J. Cardiovasc. Pharmacol. 2021, 78, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, J.F.; Pourdjabbar, A.; Quan, A.; Leong-Poi, H.; Teichert-Kuliszewska, K.; Verma, S.; Parker, T.G. Lack of S100A1 in mice confers a gender-dependent hypertensive phenotype and increased mortality after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1457–H1465. [Google Scholar] [CrossRef]
- Fan, L.; Liu, B.; Guo, R.; Luo, J.; Li, H.; Li, Z.; Xu, W. Elevated plasma S100A1 level is a risk factor for ST-segment elevation myocardial infarction and associated with post-infarction cardiac function. Int. J. Med. Sci. 2019, 16, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Sun, T.; Ping, Y.; Dai, Y.; Liu, Z.; Wang, Y.; Wang, D.; Xia, X.; Shan, H.; et al. S100A1 is a sensitive and specific cardiac biomarker for early diagnosis and prognostic assessment of acute myocardial infarction measured by chemiluminescent immunoassay. Clin. Chim. Acta 2021, 516, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Mofid, A.; Newman, N.S.; Lee, P.J.; Abbasi, C.; Matkar, P.N.; Rudenko, D.; Kuliszewski, M.A.; Chen, H.H.; Afrasiabi, K.; Tsoporis, J.N.; et al. Cardiac Overexpression of S100A6 Attenuates Cardiomyocyte Apoptosis and Reduces Infarct Size After Myocardial Ischemia-Reperfusion. J. Am. Heart Assoc. 2017, 6, e004738. [Google Scholar] [CrossRef] [PubMed]
- Doroudgar, S.; Quijada, P.; Konstandin, M.; Ilves, K.; Broughton, K.; Khalafalla, F.G.; Casillas, A.; Nguyen, K.; Gude, N.; Toko, H.; et al. S100A4 protects the myocardium against ischemic stress. J. Mol. Cell Cardiol. 2016, 100, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, A.; Laffargue, M.; Li, M.; Hirsch, E. PI3K and Calcium Signaling in Cardiovascular Disease. Circ. Res. 2017, 121, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Hong, J.; Zhang, Y.; Zhu, M.; Wang, X.; Chu, M.; Yao, J.; Xu, D. Downregulation of S100A4 Alleviates Cardiac Fibrosis via Wnt/β-Catenin Pathway in Mice. Cell Physiol. Biochem. 2018, 46, 2551–2560. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target. Ther. 2022, 7, 78. [Google Scholar] [CrossRef]
- Li, J.; Shen, D.; Tang, J.; Wang, Y.; Wang, B.; Xiao, Y.; Cao, C.; Shi, X.; Liu, H.M.; Zhao, W.; et al. IL33 attenuates ventricular remodeling after myocardial infarction through inducing alternatively activated macrophages ethical standards statement. Eur. J. Pharmacol. 2019, 854, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Veeraveedu, P.T.; Sanada, S.; Okuda, K.; Fu, H.Y.; Matsuzaki, T.; Araki, R.; Yamato, M.; Yasuda, K.; Sakata, Y.; Yoshimoto, T.; et al. Ablation of IL-33 gene exacerbate myocardial remodeling in mice with heart failure induced by mechanical stress. Biochem. Pharmacol. 2017, 138, 73–80. [Google Scholar] [CrossRef]
- Sciatti, E.; Merlo, A.; Scangiuzzi, C.; Limonta, R.; Gori, M.; D’Elia, E.; Aimo, A.; Vergaro, G.; Emdin, M.; Senni, M. Prognostic Value of sST2 in Heart Failure. J. Clin. Med. 2023, 12, 3970. [Google Scholar] [CrossRef] [PubMed]
- Witkowska, A.; Staciwa, M.; Duraj, I.; Wozniak, E.; Broncel, M.; Gorzelak-Pabis, P. Interleukin-33/sST2: Dynamic assessment in patients with acute coronary syndrome. Adv. Med. Sci. 2023, 68, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Wang, J.H.; Wang, X.L.; Liu, J.Y.; Jiang, F.Y.; Huang, X.L.; Hang, J.Y.; Qin, W.; Ma, S.X.; Zhang, J.; et al. Roles of ST2, IL-33 and BNP in predicting major adverse cardiovascular events in acute myocardial infarction after percutaneous coronary intervention. J. Cell Mol. Med. 2017, 21, 2677–2684. [Google Scholar] [CrossRef]
- Xia, J.; Qu, Y.; Yin, C.; Xu, D. Preliminary study of beta-blocker therapy on modulation of interleukin-33/ST2 signaling during ventricular remodeling after acute myocardial infarction. Cardiol. J. 2017, 24, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Geng, J.; Gao, S.X.; Yue, W.W.; Liu, Q. Eplerenone Modulates Interleukin-33/sST2 Signaling and IL-1β in Left Ventricular Systolic Dysfunction After Acute Myocardial Infarction. J. Interferon Cytokine Res. 2018, 38, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Lax, A.; Sanchez-Mas, J.; Asensio-Lopez, M.C.; Fernandez-Del Palacio, M.J.; Caballero, L.; Garrido, I.P.; Pastor-Perez, F.J.; Januzzi, J.L.; Pascual-Figal, D.A. Mineralocorticoid receptor antagonists modulate galectin-3 and interleukin-33/ST2 signaling in left ventricular systolic dysfunction after acute myocardial infarction. JACC Heart Fail. 2015, 3, 50–58. [Google Scholar] [CrossRef]
- Suica, V.I.; Uyy, E.; Ivan, L.; Boteanu, R.M.; Cerveanu-Hogas, A.; Hansen, R.; Antohe, F. Cardiac Alarmins as Residual Risk Markers of Atherosclerosis under Hypolipidemic Therapy. Int. J. Mol. Sci. 2022, 23, 11174. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cell Type | Mechanism Linking to Pathogenesis of Atherosclerosis | References |
|---|---|---|
| Vascular smooth muscle cells | HMGB1 enhances the expression of CRP and MMP-2. HMGB1 is involved in a signalling loop with NLRP3 inflammasome, as it enhances its expression. Furthermore, the inflammasome stimulates the formation of VSMC foam cells. Silencing HMGB1 potentiates the anti-inflammatory properties of other agents to suppress the synthetic phenotype of VSMC. HMGB1 is involved in the vascular calcification process. Inflammation and migration of VSMCs can be targeted using miRNAs miR-141-5p, miR-129-5p, miR-34c, and miR-24. | [18,25,26,28,31,32,33,34] |
| Endothelial cells | Ox-LDL enhances HMGB1 expression and cytoplasm translocation. Silencing HMGB1 reduces endothelial cell damage induced by modified LDL. HMGB1 enhances endoplasmic reticulum stress in endothelial cells. | [48,54] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiełbowski, K.; Skórka, P.; Plewa, P.; Bakinowska, E.; Pawlik, A. The Role of Alarmins in the Pathogenesis of Atherosclerosis and Myocardial Infarction. Curr. Issues Mol. Biol. 2024, 46, 8995-9015. https://doi.org/10.3390/cimb46080532
Kiełbowski K, Skórka P, Plewa P, Bakinowska E, Pawlik A. The Role of Alarmins in the Pathogenesis of Atherosclerosis and Myocardial Infarction. Current Issues in Molecular Biology. 2024; 46(8):8995-9015. https://doi.org/10.3390/cimb46080532
Chicago/Turabian StyleKiełbowski, Kajetan, Patryk Skórka, Paulina Plewa, Estera Bakinowska, and Andrzej Pawlik. 2024. "The Role of Alarmins in the Pathogenesis of Atherosclerosis and Myocardial Infarction" Current Issues in Molecular Biology 46, no. 8: 8995-9015. https://doi.org/10.3390/cimb46080532