
Unlocking the Genetic Secrets of Acromegaly: Exploring the Role of Genetics in a Rare Disorder

 ,
, 
Abstract
:
1. Introduction
2. Isolated Acromegaly
2.1. Familial Isolated Pituitary Adenomas—FIPAs
2.1.1. Aryl Hydrocarbon Receptor-Interacting Protein (AIP)
2.1.2. G Protein-Coupled Receptor 101 (GPR101)–X-Linked Acrogigantism (X-LAG)
2.1.3. Unknown Mutations
2.2. Sporadic Acromegaly
2.2.1. AIP
2.2.2. GNAS Complex Locus (GNAS)
2.2.3. Higher Gastric Inhibitory Polypeptide Receptor (GIPR) Expression
3. Syndromic Acromegaly
3.1. Multiple Endocrine Neoplasia Type 1 (MEN1)
3.2. Multiple Endocrine Neoplasia Type 4 (MEN4)
3.3. Carney Complex (CNC)
3.4. McCune–Albright Syndrome (MAS)
3.5. 3P Association (3Pa)
3.6. Neurofibromatosis Type 1 (NF1)
3.7. Tuberous Sclerosis Complex (TSC)
3.8. Multiple Endocrine Neoplasia Type 2 (MEN2)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Asa, S.L.; Casar-Borota, O.; Chanson, P.; Delgrange, E.; Earls, P.; Ezzat, S.; Grossman, A.; Ikeda, H.; Inoshita, N.; Karavitaki, N.; et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): An International Pituitary Pathology Club proposal. Endocr. Relat. Cancer 2017, 24, C5–C8. [Google Scholar] [CrossRef] [PubMed]
- AlDallal, S. Acromegaly: A challenging condition to diagnose. Int. J. Gen. Med. 2018, 11, 337–343. [Google Scholar] [CrossRef]
- Boguslawska, A.; Korbonits, M. Genetics of Acromegaly and Gigantism. J. Clin. Med. 2021, 10, 1377. [Google Scholar] [CrossRef]
- Dineen, R.; Stewart, P.M.; Sherlock, M. Acromegaly. Int. J. Med. 2017, 110, 411–420. [Google Scholar] [CrossRef]
- Crisafulli, S.; Luxi, N.; Sultana, J.; Fontana, A.; Spagnolo, F.; Giuffrida, G.; Ferrau, F.; Gianfrilli, D.; Cozzolino, A.; Cristina De Martino, M.; et al. Global epidemiology of acromegaly: A systematic review and meta-analysis. Eur. J. Endocrinol. 2021, 185, 251–263. [Google Scholar] [CrossRef]
- Vilar, L.; Vilar, C.F.; Lyra, R.; Lyra, R.; Naves, L.A. Acromegaly: Clinical features at diagnosis. Pituitary 2017, 20, 22–32. [Google Scholar] [CrossRef]
- Esposito, D.; Ragnarsson, O.; Johannsson, G.; Olsson, D.S. Prolonged diagnostic delay in acromegaly is associated with increased morbidity and mortality. Eur. J. Endocrinol. 2020, 182, 523–531. [Google Scholar] [CrossRef]
- Bolfi, F.; Neves, A.F.; Boguszewski, C.L.; Nunes-Nogueira, V.S. Mortality in acromegaly decreased in the last decade: A systematic review and meta-analysis. Eur. J. Endocrinol. 2018, 179, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Gliga, M.C.; Tataranu, L.G.; Popescu, M.; Chinezu, L.; Pascanu, M.I. Immunohistochemical evaluation of biomarkers with predictive role in acromegaly: A literature review. Rom. J. Morphol. Embryol. 2023, 64, 25–33. [Google Scholar] [CrossRef]
- UniProt. UniProt: The Universal Protein Knowledgebase. Available online: https://www.uniprot.org/ (accessed on 14 April 2024).
- Ershadinia, N.; Tritos, N.A. Diagnosis and Treatment of Acromegaly: An Update. Mayo Clin. Proc. 2022, 97, 333–346. [Google Scholar] [CrossRef]
- Banerjee, A.; Goel, A.; Shah, A.; Srivastava, S. Recent advances in proteomics and its implications in pituitary endocrine disorders. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140700. [Google Scholar] [CrossRef]
- Miles, B.; Tadi, P. Genetics, Somatic Mutation; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Stratakis, C.A.; Tichomirowa, M.A.; Boikos, S.; Azevedo, M.F.; Lodish, M.; Martari, M.; Verma, S.; Daly, A.F.; Raygada, M.; Keil, M.F.; et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clin. Genet. 2010, 78, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Beckers, A.; Aaltonen, L.A.; Daly, A.F.; Karhu, A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr. Rev. 2013, 34, 239–277. [Google Scholar] [CrossRef]
- Marques, P.; Caimari, F.; Hernandez-Ramirez, L.C.; Collier, D.; Iacovazzo, D.; Ronaldson, A.; Magid, K.; Lim, C.T.; Stals, K.; Ellard, S.; et al. Significant Benefits of AIP Testing and Clinical Screening in Familial Isolated and Young-onset Pituitary Tumors. J. Clin. Endocrinol. Metab. 2020, 105, e2247–e2260. [Google Scholar] [CrossRef]
- Korbonits, M.; Kumar, A.V. AIP Familial Isolated Pituitary Adenomas. In GeneReviews® [Internet]; [Updated 2020]; University of Washington: Seattle, WA, USA, 2012. [Google Scholar]
- Daly, A.F.; Tichomirowa, M.A.; Petrossians, P.; Heliovaara, E.; Jaffrain-Rea, M.L.; Barlier, A.; Naves, L.A.; Ebeling, T.; Karhu, A.; Raappana, A.; et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: An international collaborative study. J. Clin. Endocrinol. Metab. 2010, 95, E373–E383. [Google Scholar] [CrossRef] [PubMed]
- Leontiou, C.A.; Gueorguiev, M.; van der Spuy, J.; Quinton, R.; Lolli, F.; Hassan, S.; Chahal, H.S.; Igreja, S.C.; Jordan, S.; Rowe, J.; et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J. Clin. Endocrinol. Metab. 2008, 93, 2390–2401. [Google Scholar] [CrossRef]
- Vasilev, V.; Daly, A.F.; Trivellin, G.; Stratakis, C.A.; Zacharieva, S.; Beckers, A. HEREDITARY ENDOCRINE TUMOURS: CURRENT STATE-OF-THE-ART AND RESEARCH OPPORTUNITIES: The roles of AIP and GPR101 in familial isolated pituitary adenomas (FIPA). Endocr. Relat. Cancer 2020, 27, T77–T86. [Google Scholar] [CrossRef]
- Vasilev, V.; Daly, A.F.; Zacharieva, S.; Beckers, A. Clinical and Molecular Update on Genetic Causes of Pituitary Adenomas. Horm. Metab. Res. 2020, 52, 553–561. [Google Scholar] [CrossRef] [PubMed]
- HGNC. HGNC: HUGO Gene Nomenclature Committee Home Page. Available online: https://www.genenames.org/ (accessed on 14 April 2024).
- Igreja, S.; Chahal, H.S.; King, P.; Bolger, G.B.; Srirangalingam, U.; Guasti, L.; Chapple, J.P.; Trivellin, G.; Gueorguiev, M.; Guegan, K.; et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum. Mutat. 2010, 31, 950–960. [Google Scholar] [CrossRef]
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.H.; et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N. Engl. J. Med. 2014, 371, 2363–2374. [Google Scholar] [CrossRef]
- Iacovazzo, D.; Caswell, R.; Bunce, B.; Jose, S.; Yuan, B.; Hernandez-Ramirez, L.C.; Kapur, S.; Caimari, F.; Evanson, J.; Ferrau, F.; et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: A clinico-pathological and genetic study. Acta Neuropathol. Commun. 2016, 4, 56. [Google Scholar] [CrossRef]
- Beckers, A.; Lodish, M.B.; Trivellin, G.; Rostomyan, L.; Lee, M.; Faucz, F.R.; Yuan, B.; Choong, C.S.; Caberg, J.H.; Verrua, E.; et al. X-linked acrogigantism syndrome: Clinical profile and therapeutic responses. Endocr. Relat. Cancer 2015, 22, 353–367. [Google Scholar] [CrossRef]
- Iacovazzo, D.; Korbonits, M. X-Linked Acrogigantism. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Alzahrani, A.S.; Bin Nafisah, A.; Alswailem, M.; Alghamdi, B.; Alsaihati, B.; Aljafar, H.; Baz, B.; Alhindi, H.; Moria, Y.; Butt, M.I.; et al. Germline Variants in Sporadic Pituitary Adenomas. J. Endocr. Soc. 2024, 8, bvae085. [Google Scholar] [CrossRef]
- Srirangam Nadhamuni, V.; Korbonits, M. Novel Insights into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms. Endocr. Rev. 2020, 41, 821–846. [Google Scholar] [CrossRef]
- Trivellin, G.; Daly, A.F.; Hernandez-Ramirez, L.C.; Araldi, E.; Tatsi, C.; Dale, R.K.; Fridell, G.; Mittal, A.; Faucz, F.R.; Iben, J.R.; et al. Germline loss-of-function PAM variants are enriched in subjects with pituitary hypersecretion. Front. Endocrinol. 2023, 14, 1166076. [Google Scholar] [CrossRef]
- Cazabat, L.; Bouligand, J.; Salenave, S.; Bernier, M.; Gaillard, S.; Parker, F.; Young, J.; Guiochon-Mantel, A.; Chanson, P. Germline AIP mutations in apparently sporadic pituitary adenomas: Prevalence in a prospective single-center cohort of 443 patients. J. Clin. Endocrinol. Metab. 2012, 97, E663–E670. [Google Scholar] [CrossRef]
- Gaspar, L.M.; Goncalves, C.I.; Saraiva, C.; Cortez, L.; Amaral, C.; Nobre, E.; Lemos, M.C. Low frequency of AIP mutations in patients with young-onset sporadic pituitary macroadenomas. J. Endocrinol. Investig. 2023, 46, 2299–2307. [Google Scholar] [CrossRef]
- Oriola, J.; Lucas, T.; Halperin, I.; Mora, M.; Perales, M.J.; Alvarez-Escola, C.; de Paz, M.N.; Diaz Soto, G.; Salinas, I.; Julian, M.T.; et al. Germline mutations of AIP gene in somatotropinomas resistant to somatostatin analogues. Eur. J. Endocrinol. 2013, 168, 9–13. [Google Scholar] [CrossRef]
- Daly, A.F.; Tichomirowa, M.A.; Beckers, A. The epidemiology and genetics of pituitary adenomas. Best. Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 543–554. [Google Scholar] [CrossRef]
- Ronchi, C.L.; Peverelli, E.; Herterich, S.; Weigand, I.; Mantovani, G.; Schwarzmayr, T.; Sbiera, S.; Allolio, B.; Honegger, J.; Appenzeller, S.; et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2016, 174, 363–372. [Google Scholar] [CrossRef]
- Han, S.R.; Lee, Y.A.; Shin, C.H.; Yang, S.W.; Lim, B.C.; Cho, T.J.; Ko, J.M. Clinical and Molecular Characteristics of GNAS Inactivation Disorders Observed in 18 Korean Patients. Exp. Clin. Endocrinol. Diabetes 2021, 129, 118–125. [Google Scholar] [CrossRef]
- Agrawal, N.; Gersey, Z.C.; Abou-Al-Shaar, H.; Gardner, P.A.; Mantica, M.; Agnihotri, S.; Mahmud, H.; Fazeli, P.K.; Zenonos, G.A. Major Genetic Motifs in Pituitary Adenomas: A Practical Literature Update. World Neurosurg. 2023, 169, 43–50. [Google Scholar] [CrossRef]
- Freda, P.U.; Chung, W.K.; Matsuoka, N.; Walsh, J.E.; Kanibir, M.N.; Kleinman, G.; Wang, Y.; Bruce, J.N.; Post, K.D. Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: Correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary 2007, 10, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, K.; Kim, D.; Moon, J.H.; Kim, E.H.; Kim, S.H.; Ku, C.R.; Lee, E.J. Associations of GNAS Mutations with Surgical Outcomes in Patients with Growth Hormone-Secreting Pituitary Adenoma. Endocrinol. Metab. 2021, 36, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ramirez, L.C. Potential markers of disease behavior in acromegaly and gigantism. Expert. Rev. Endocrinol. Metab. 2020, 15, 171–183. [Google Scholar] [CrossRef]
- Rymuza, J.; Kober, P.; Rusetska, N.; Mossakowska, B.J.; Maksymowicz, M.; Nyc, A.; Baluszek, S.; Zielinski, G.; Kunicki, J.; Bujko, M. Transcriptomic Classification of Pituitary Neuroendocrine Tumors Causing Acromegaly. Cells 2022, 11, 3846. [Google Scholar] [CrossRef]
- Neou, M.; Villa, C.; Armignacco, R.; Jouinot, A.; Raffin-Sanson, M.L.; Septier, A.; Letourneur, F.; Diry, S.; Diedisheim, M.; Izac, B.; et al. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell 2020, 37, 123–134. [Google Scholar] [CrossRef]
- Peverelli, E.; Treppiedi, D.; Mangili, F.; Catalano, R.; Mantovani, G. Chapter 10—GNAS, McCune–Albright syndrome, and GH-producing tumors. In Gigantism and Acromegaly; Stratakis, C.A., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 197–223. [Google Scholar] [CrossRef]
- Amazit, L.; Barbot, M.; Beau, I.; Bouligand, J.; Bourdeau, I.; Chanson, P.; Cloix, L.; Corbeil, G.; de Herder, W.; Deméocq, V.; et al. OR04-4 Loss of KDM1A in Bilateral Macronodular Adrenal Hyperplasia With GIP-Dependent Cushing’s Syndrome and in Acromegaly With Paradoxical GH Response to Oral Glucose. J. Endocr. Soc. 2022, 6, A81. [Google Scholar] [CrossRef]
- Chasseloup, F.; Regazzo, D.; Tosca, L.; Proust, A.; Kuhn, E.; Hage, M.; Jublanc, C.; Mokhtari, K.; Dalle Nogare, M.; Avallone, S.; et al. KDM1A genotyping and expression in 146 sporadic somatotroph pituitary adenomas. Eur. J. Endocrinol. 2024, 190, 173–181. [Google Scholar] [CrossRef]
- Hage, M.; Janot, C.; Salenave, S.; Chanson, P.; Kamenický, P. MANAGEMENT OF ENDOCRINE DISEASE: Etiology and outcome of acromegaly in patients with a paradoxical GH response to glucose. Eur. J. Endocrinol. 2021, 184, R261–R268. [Google Scholar] [CrossRef]
- Giusti, F.; Marini, F.; Brandi, M.L. Multiple Endocrine Neoplasia Type 1. In GeneReviews® [Internet]; [Updated 2022]; University of Washington: Seattle, WA, USA, 2005. [Google Scholar]
- Al-Salameh, A.; Cadiot, G.; Calender, A.; Goudet, P.; Chanson, P. Clinical aspects of multiple endocrine neoplasia type 1. Nat. Rev. Endocrinol. 2021, 17, 207–224. [Google Scholar] [CrossRef]
- Goudet, P.; Cadiot, G.; Barlier, A.; Baudin, E.; Borson-Chazot, F.; Brunaud, L.; Caiazzo, R.; Cardot-Bauters, C.; Castinetti, F.; Chanson, P.; et al. French guidelines from the GTE, AFCE and ENDOCAN-RENATEN (Groupe d’etude des Tumeurs Endocrines/Association Francophone de Chirurgie Endocrinienne/Reseau national de prise en charge des tumeurs endocrines) for the screening, diagnosis and management of Multiple Endocrine Neoplasia Type 1. Ann. Endocrinol. 2024, 85, 2–19. [Google Scholar] [CrossRef]
- Nachtigall, L.B.; Guarda, F.J.; Lines, K.E.; Ghajar, A.; Dichtel, L.; Mumbach, G.; Zhao, W.; Zhang, X.; Tritos, N.A.; Swearingen, B.; et al. Clinical MEN-1 among a Large Cohort of Patients With Acromegaly. J. Clin. Endocrinol. Metab. 2020, 105, e2271–e2281. [Google Scholar] [CrossRef]
- Verges, B.; Boureille, F.; Goudet, P.; Murat, A.; Beckers, A.; Sassolas, G.; Cougard, P.; Chambe, B.; Montvernay, C.; Calender, A. Pituitary disease in MEN type 1 (MEN1): Data from the France-Belgium MEN1 multicenter study. J. Clin. Endocrinol. Metab. 2002, 87, 457–465. [Google Scholar] [CrossRef]
- Mele, C.; Mencarelli, M.; Caputo, M.; Mai, S.; Pagano, L.; Aimaretti, G.; Scacchi, M.; Falchetti, A.; Marzullo, P. Phenotypes Associated With MEN1 Syndrome: A Focus on Genotype-Phenotype Correlations. Front. Endocrinol. 2020, 11, 591501. [Google Scholar] [CrossRef]
- NIH. NIH: National Library of Medicine; Gene ID: 4221. Available online: https://www.ncbi.nlm.nih.gov/gene/4221#gene-expression (accessed on 18 April 2024).
- OMIM. Online Mendelian Inheritance in Man (OMIM). Available online: https://www.omim.org/ (accessed on 18 April 2024).
- Brandi, M.L.; Agarwal, S.K.; Perrier, N.D.; Lines, K.E.; Valk, G.D.; Thakker, R.V. Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr. Rev. 2021, 42, 133–170. [Google Scholar] [CrossRef]
- Nelakurti, D.D.; Pappula, A.L.; Rajasekaran, S.; Miles, W.O.; Petreaca, R.C. Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer. Cancers 2020, 12, 2616. [Google Scholar] [CrossRef]
- Marques, P.; Magalhães, D.; Caimari, F.; Hernández Ramírez, L.; Collier, D.; Lim, C.T.; Stals, K.; Ellard, S.; Druce, M.; Akker, S.; et al. Phenotypic differences between patients with familial pituitary neuroendocrine tumours due to MEN1 or AIP mutations. Endocr. Abstr. 2020, 70, AEP600. [Google Scholar] [CrossRef]
- Brauer, V.F.; Scholz, G.H.; Neumann, S.; Lohmann, T.; Paschke, R.; Koch, C.A. RET germline mutation in codon 791 in a family representing 3 generations from age 5 to age 70 years: Should thyroidectomy be performed? Endocr. Pract. 2004, 10, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Chiloiro, S.; Capoluongo, E.D.; Costanza, F.; Minucci, A.; Giampietro, A.; Infante, A.; Milardi, D.; Ricciardi Tenore, C.; De Bonis, M.; Gaudino, S.; et al. The Pathogenic RET Val804Met Variant in Acromegaly: A New Clinical Phenotype? Int. J. Mol. Sci. 2024, 25, 1895. [Google Scholar] [CrossRef]
- Saito, T.; Miura, D.; Taguchi, M.; Takeshita, A.; Miyakawa, M.; Takeuchi, Y. Coincidence of multiple endocrine neoplasia type 2A with acromegaly. Am. J. Med. Sci. 2010, 340, 329–331. [Google Scholar] [CrossRef]
- Yasir, M.; Mulji, N.J.; Kasi, A. Multiple Endocrine Neoplasias Type 2; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Alrezk, R.; Hannah-Shmouni, F.; Stratakis, C.A. MEN4 and CDKN1B mutations: The latest of the MEN syndromes. Endocr. Relat. Cancer 2017, 24, T195–T208. [Google Scholar] [CrossRef]
- Ruggeri, R.M.; Benevento, E.; De Cicco, F.; Grossrubatscher, E.M.; Hasballa, I.; Tarsitano, M.G.; Centello, R.; Isidori, A.M.; Colao, A.; Pellegata, N.S.; et al. Multiple endocrine neoplasia type 4 (MEN4): A thorough update on the latest and least known men syndrome. Endocrine 2023, 82, 480–490. [Google Scholar] [CrossRef]
- Frederiksen, A.; Rossing, M.; Hermann, P.; Ejersted, C.; Thakker, R.V.; Frost, M. Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases. J. Clin. Endocrinol. Metab. 2019, 104, 3637–3646. [Google Scholar] [CrossRef]
- Brock, P.; Kirschner, L. Multiple Endocrine Neoplasia Type 4. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2023. [Google Scholar]
- Bouys, L.; Bertherat, J. MANAGEMENT OF ENDOCRINE DISEASE: Carney complex: Clinical and genetic update 20 years after the identification of the CNC1 (PRKAR1A) gene. Eur. J. Endocrinol. 2021, 184, R99–R109. [Google Scholar] [CrossRef] [PubMed]
- González, R.G.; Padilla, A.M.A.; Romero, J.A.O.; Verdugo, M.A.C. Carney Complex: A Comprehensive Review of Clinical Presentation, Genetics, and Therapeutic Advances. Int. J. Med. Sci. Clin. Res. Stud. 2024, 4, 30–35. [Google Scholar] [CrossRef]
- Nagata, T.; Kawano, A.; Koyama, M.; Nakamura, T.; Hirahara, F.; Nakajima, T.; Sato, T.; Sakakibara, H. Efficacy of Fibroblast Growth Factor on Epithelialization of the Neovagina in Patients with Mayer-Rokitansky-Kuster-Hauser Syndrome Who Underwent Vaginoplasty. J. Pediatr. Adolesc. Gynecol. 2017, 30, 400–404. [Google Scholar] [CrossRef]
- Cuny, T.; Mac, T.T.; Romanet, P.; Dufour, H.; Morange, I.; Albarel, F.; Lagarde, A.; Castinetti, F.; Graillon, T.; North, M.O.; et al. Acromegaly in Carney complex. Pituitary 2019, 22, 456–466. [Google Scholar] [CrossRef]
- Kamilaris, C.D.C.; Faucz, F.R.; Voutetakis, A.; Stratakis, C.A. Carney Complex. Exp. Clin. Endocrinol. Diabetes 2019, 127, 156–164. [Google Scholar] [CrossRef] [PubMed]
- NIH. National Library of Medicine; Gene ID: 5573. Available online: https://www.ncbi.nlm.nih.gov/gene/5573 (accessed on 17 April 2024).
- Stratakis, C.A. Carney Complex. In GeneReviews® [Internet]; [Updated 2023]; University of Washington: Seattle, WA, USA, 2003. [Google Scholar]
- Safarinejad, M.R. Expression of Concern: Evaluation of Semen Quality, Endocrine Profile and Hypothalamus-Pituitary-Testis Axis in Male Patients With Homozygous beta-Thalassemia Major. J. Urol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Liu, Y.; Wang, L.; Deng, K.; Yang, H.; Lu, L.; Feng, F.; Xing, B.; You, H.; Jin, Z.; et al. Clinical characteristics and management of growth hormone excess in patients with McCune-Albright syndrome. Eur. J. Endocrinol. 2017, 176, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Szymczuk, V.; Florenzano, P.; de Castro, L.F.; Collins, M.T.; Boyce, A.M. Fibrous Dysplasia/McCune-Albright Syndrome. In GeneReviews® [Internet]; [Updated 2024]; University of Washington: Seattle, WA, USA, 2015. [Google Scholar]
- Ilie, M.-D.; Raverot, G.; Brac de la Perrière, A. Pasireotide: Potential treatment option for McCune–Albright-associated acromegaly. Eur. J. Endocrinol. 2023, 190, K17–K20. [Google Scholar] [CrossRef] [PubMed]
- Mougel, G.; Lagarde, A.; Albarel, F.; Essamet, W.; Luigi, P.; Mouly, C.; Vialon, M.; Cuny, T.; Castinetti, F.; Saveanu, A.; et al. Germinal defects of SDHx genes in patients with isolated pituitary adenoma. Eur. J. Endocrinol. 2020, 183, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.; Korbonits, M. Pituitary Dysfunction in Systemic Disorders. In The Pituitary; Melmed, S., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2022; pp. 385–412. [Google Scholar]
- Else, T.; Greenberg, S.; Fishbein, L. Hereditary Paraganglioma-Pheochromocytoma Syndromes. In GeneReviews® [Internet]; [Updated 2023]; University of Washington: Seattle, WA, USA, 2008. [Google Scholar]
- Friedman, J.M. Neurofibromatosis 1. In GeneReviews® [Internet]; [Updated 2022]; University of Washington: Seattle, WA, USA, 1998. [Google Scholar]
- Yasuda, S.; Inoue, I.; Shimada, A. Neurofibromatosis Type 1 with Concurrent Multiple Endocrine Disorders: Adenomatous Goiter, Primary Hyperparathyroidism, and Acromegaly. Intern. Med. 2021, 60, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Hannah-Shmouni, F.; Trivellin, G.; Beckers, P.; Karaviti, L.P.; Lodish, M.; Tatsi, C.; Adesina, A.M.; Adamidou, F.; Mintziori, G.; Josefson, J.L.; et al. Neurofibromatosis Type 1 Has a Wide Spectrum of Growth Hormone Excess. J. Clin. Med. 2022, 11, 2168. [Google Scholar] [CrossRef]
- Cambiaso, P.; Galassi, S.; Palmiero, M.; Mastronuzzi, A.; Del Bufalo, F.; Capolino, R.; Cacchione, A.; Buonuomo, P.S.; Gonfiantini, M.V.; Bartuli, A.; et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am. J. Med. Genet. A 2017, 173, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Peduto, C.; Zanobio, M.; Nigro, V.; Perrotta, S.; Piluso, G.; Santoro, C. Neurofibromatosis Type 1: Pediatric Aspects and Review of Genotype-Phenotype Correlations. Cancers 2023, 15, 1217. [Google Scholar] [CrossRef] [PubMed]
- NIH. National Library of Medicine; Gene ID: 4763. Available online: https://www.ncbi.nlm.nih.gov/gene/4763 (accessed on 15 April 2024).
- Bruzzi, P.; Sani, I.; Albanese, A. Reversible Growth Hormone Excess in Two Girls with Neurofibromatosis Type 1 and Optic Pathway Glioma. Horm. Res. Paediatr. 2015, 84, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Galaction-Nitelea, O.; Dociu, I.; Murgu, V. A case of tuberous sclerosis with acromegaly. Rev. Med. Interna Neurol. Psihiatr. Neurochir. Dermatovenerol. Neurol. Psihiatr. Neurochir. 1978, 23, 253–262. [Google Scholar]
- Hoffman, W.H.; Perrin, J.C.; Halac, E.; Gala, R.R.; England, B.G. Acromegalic gigantism and tuberous sclerosis. J. Pediatr. 1978, 93, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Peron, A.; Au, K.S.; Northrup, H. Genetics, genomics, and genotype-phenotype correlations of TSC: Insights for clinical practice. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Hernández Ramírez, L. Chapter 6—The role of the aryl hydrocarbon receptor interacting protein in pituitary tumorigenesis. In Gigantism and Acromegaly; Stratakis, C.A., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 89–126. [Google Scholar] [CrossRef]
- Daly, A.F.; Beckers, A. Familial isolated pituitary adenomas (FIPA) and mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocrinol. Metab. Clin. North. Am. 2015, 44, 19–25. [Google Scholar] [CrossRef]
- Cain, J.W.; Miljic, D.; Popovic, V.; Korbonits, M. Role of the aryl hydrocarbon receptor-interacting protein in familial isolated pituitary adenoma. Expert. Rev. Endocrinol. Metab. 2010, 5, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Coopmans, E.C.; Korbonits, M. Molecular genetic testing in the management of pituitary disease. Clin. Endocrinol. 2022, 97, 424–435. [Google Scholar] [CrossRef]
- NIH. NIH: National Library of Medicine; Gene ID: 9049. Available online: https://www.ncbi.nlm.nih.gov/gene/9049 (accessed on 14 April 2024).
- Formosa, R.; Vassallo, J. Aryl Hydrocarbon Receptor-Interacting Protein (AIP) N-Terminus Gene Mutations Identified in Pituitary Adenoma Patients Alter Protein Stability and Function. Horm. Cancer 2017, 8, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Schernthaner-Reiter, M.H.; Trivellin, G.; Stratakis, C.A. Interaction of AIP with protein kinase A (cAMP-dependent protein kinase). Hum. Mol. Genet. 2018, 27, 2604–2613. [Google Scholar] [CrossRef]
- VarSome. VarSome: The Human Genomic Variant Search Engine. Available online: https://varsome.com/ (accessed on 14 April 2024).
- Tichomirowa, M.A.; Barlier, A.; Daly, A.F.; Jaffrain-Rea, M.L.; Ronchi, C.; Yaneva, M.; Urban, J.D.; Petrossians, P.; Elenkova, A.; Tabarin, A.; et al. High prevalence of AIP gene mutations following focused screening in young patients with sporadic pituitary macroadenomas. Eur. J. Endocrinol. 2011, 165, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.; Hunter, S.; Bradley, L.; Chahal, H.S.; Storr, H.L.; Akker, S.A.; Kumar, A.V.; Orme, S.M.; Evanson, J.; Abid, N.; et al. Clinical experience in the screening and management of a large kindred with familial isolated pituitary adenoma due to an aryl hydrocarbon receptor interacting protein (AIP) mutation. J. Clin. Endocrinol. Metab. 2014, 99, 1122–1131. [Google Scholar] [CrossRef]
- Korbonits, M.; Storr, H.; Kumar, A.V. Familial pituitary adenomas—Who should be tested for AIP mutations? Clin. Endocrinol. 2012, 77, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Beckers, A. The Genetic Pathophysiology and Clinical Management of the TADopathy, X-Linked Acrogigantism. Endocr. Rev. 2024, bnae014. [Google Scholar] [CrossRef]
- Caruso, M.; Mazzatenta, D.; Asioli, S.; Costanza, G.; Trivellin, G.; Franke, M.; Abboud, D.; Hanson, J.; Raverot, V.; Petrossians, P.; et al. Case report: Management of pediatric gigantism caused by the TADopathy, X-linked acrogigantism. Front. Endocrinol. 2024, 15, 1345363. [Google Scholar] [CrossRef]
- Pepe, S.; Korbonits, M.; Iacovazzo, D. Germline and mosaic mutations causing pituitary tumours: Genetic and molecular aspects. J. Endocrinol. 2019, 240, R21–R45. [Google Scholar] [CrossRef]
- Abboud, D.; Daly, A.F.; Dupuis, N.; Bahri, M.A.; Inoue, A.; Chevigne, A.; Ectors, F.; Plenevaux, A.; Pirotte, B.; Beckers, A.; et al. GPR101 drives growth hormone hypersecretion and gigantism in mice via constitutive activation of G(s) and G(q/11). Nat. Commun. 2020, 11, 4752. [Google Scholar] [CrossRef] [PubMed]
- Costanzi, S.; Stahr, L.G.; Trivellin, G.; Stratakis, C.A. GPR101: Modeling a constitutively active receptor linked to X-linked acrogigantism. J. Mol. Graph. Model. 2024, 127, 108676. [Google Scholar] [CrossRef] [PubMed]
- Beckers, A.; Fernandes, D.; Fina, F.; Novak, M.; Abati, A.; Rostomyan, L.; Thiry, A.; Ouafik, L.; Pasture, B.; Pinhasi, R.; et al. Paleogenetic study of ancient DNA suggestive of X-linked acrogigantism. Endocr. Relat. Cancer 2017, 24, L17–L20. [Google Scholar] [CrossRef]
- Rodd, C.; Millette, M.; Iacovazzo, D.; Stiles, C.E.; Barry, S.; Evanson, J.; Albrecht, S.; Caswell, R.; Bunce, B.; Jose, S.; et al. Somatic GPR101 Duplication Causing X-Linked Acrogigantism (XLAG)-Diagnosis and Management. J. Clin. Endocrinol. Metab. 2016, 101, 1927–1930. [Google Scholar] [CrossRef]
- Gordon, R.J.; Bell, J.; Chung, W.K.; David, R.; Oberfield, S.E.; Wardlaw, S.L. Childhood acromegaly due to X-linked acrogigantism: Long term follow-up. Pituitary 2016, 19, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Yuan, B.; Fina, F.; Caberg, J.H.; Trivellin, G.; Rostomyan, L.; de Herder, W.W.; Naves, L.A.; Metzger, D.; Cuny, T.; et al. Somatic mosaicism underlies X-linked acrogigantism syndrome in sporadic male subjects. Endocr. Relat. Cancer 2016, 23, 221–233. [Google Scholar] [CrossRef]
- Daly, A.F.; Lysy, P.A.; Desfilles, C.; Rostomyan, L.; Mohamed, A.; Caberg, J.H.; Raverot, V.; Castermans, E.; Marbaix, E.; Maiter, D.; et al. GHRH excess and blockade in X-LAG syndrome. Endocr. Relat. Cancer 2016, 23, 161–170. [Google Scholar] [CrossRef]
- Naves, L.A.; Daly, A.F.; Dias, L.A.; Yuan, B.; Zakir, J.C.; Barra, G.B.; Palmeira, L.; Villa, C.; Trivellin, G.; Junior, A.J.; et al. Aggressive tumor growth and clinical evolution in a patient with X-linked acro-gigantism syndrome. Endocrine 2016, 51, 236–244. [Google Scholar] [CrossRef]
- Rostomyan, L.; Daly, A.F.; Petrossians, P.; Nachev, E.; Lila, A.R.; Lecoq, A.L.; Lecumberri, B.; Trivellin, G.; Salvatori, R.; Moraitis, A.G.; et al. Clinical and genetic characterization of pituitary gigantism: An international collaborative study in 208 patients. Endocr. Relat. Cancer 2015, 22, 745–757. [Google Scholar] [CrossRef]
- Trivellin, G.; Hernandez-Ramirez, L.C.; Swan, J.; Stratakis, C.A. An orphan G-protein-coupled receptor causes human gigantism and/or acromegaly: Molecular biology and clinical correlations. Best. Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Akkus, G.; Korbonits, M. Genetic Testing in Hereditary Pituitary Tumors. Arch. Med. Res. 2023, 54, 102920. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.J.; Reitman, Z.J.; Ma, Z.Y.; Chen, J.H.; Zhang, Q.L.; Shou, X.F.; Huang, C.X.; Wang, Y.F.; Li, S.Q.; Mao, Y.; et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. 2016, 26, 1255–1259. [Google Scholar] [CrossRef]
- Shi, Y.; Tang, D.; Deng, J.; Su, C. Detection of gsp oncogene in growth hormone-secreting pituitary adenomas and the study of clinical characteristics of acromegalic patients with gsp-positive pituitary tumors. Chin. Med. J. 1998, 111, 891–894. [Google Scholar]
- Zhang, F.; Zhang, Q.; Zhu, J.; Yao, B.; Ma, C.; Qiao, N.; He, S.; Ye, Z.; Wang, Y.; Han, R.; et al. Integrated proteogenomic characterization across major histological types of pituitary neuroendocrine tumors. Cell Res. 2022, 32, 1047–1067. [Google Scholar] [CrossRef] [PubMed]
- Ceccato, F.; Vedolin, C.K.; Voltan, G.; Antonelli, G.; Barbot, M.; Basso, D.; Regazzo, D.; Scaroni, C.; Occhi, G. Paradoxical GH increase after oral glucose load in subjects with and without acromegaly. J. Endocrinol. Investig. 2024, 47, 213–221. [Google Scholar] [CrossRef]
- Duger, H.; Bostan, H.; Deryol, H.Y.; Imga, N.N.; Ucan, B.; Calapkulu, M.; Hepsen, S.; Akhanli, P.; Gul, U.; Sencar, M.E.; et al. Paradoxical GH increase during oral glucose load may predict overall remission in acromegalic patients. Growth Horm. IGF Res. 2022, 67, 101501. [Google Scholar] [CrossRef]
- Hage, M.; Chaligné, R.; Viengchareun, S.; Villa, C.; Salenave, S.; Bouligand, J.; Letouzé, E.; Tosca, L.; Rouquette, A.; Tachdjian, G.; et al. Hypermethylator Phenotype and Ectopic GIP Receptor in GNAS Mutation-Negative Somatotropinomas. J. Clin. Endocrinol. Metab. 2019, 104, 1777–1787. [Google Scholar] [CrossRef]
- Chasseloup, F.; Tosca, L.; Regazzo, D.; Proust, A.; Hage, M.; Kuhn, E.; Jublanc, C.; Mokhtari, K.; Salenave, S.; Gaillard, S.; et al. OR17-05 Lysine Demethylase KDM1A and Ectopic Expression of GIP-Receptor in Somatotropinomas of Patients with Paradoxical Response to Oral Glucose. J. Endocr. Soc. 2023, 7, bvad114.1310. [Google Scholar] [CrossRef]
- Dalle Nogare, M.; D’Annunzio, S.; Vazza, G.; Regazzo, D.; Picello, L.; Denaro, L.; Voltan, G.; Scaroni, C.; Ceccato, F.; Occhi, G. The Methylation Analysis of the Glucose-Dependent Insulinotropic Polypeptide Receptor (GIPR) Locus in GH-Secreting Pituitary Adenomas. Int. J. Mol. Sci. 2023, 24, 9264. [Google Scholar] [CrossRef]
- Losa, M.; Garbin, E.; Calcagnile, R.; Voltan, G.; Ceccato, F.; Scaroni, C.; Occhi, G.; Mortini, P. The Preoperative Paradoxical GH Response to Oral Glucose Load Predicts a low Risk of Recurrence in Acromegaly. J. Clin. Endocrinol. Metab. 2024, dgae410. [Google Scholar] [CrossRef]
- Kamilaris, C.D.C.; Stratakis, C.A. Multiple Endocrine Neoplasia Type 1 (MEN1): An Update and the Significance of Early Genetic and Clinical Diagnosis. Front. Endocrinol. 2019, 10, 339. [Google Scholar] [CrossRef]
- Yarman, S.; Tuncer, F.N.; Serbest, E. Three Novel MEN1 Variants in AIP-Negative Familial Isolated Pituitary Adenoma Patients. Pathobiology 2019, 86, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; Melmed, S.; Sakurai, A.; Tonelli, F.; Brandi, M.L.; et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J. Clin. Endocrinol. Metab. 2012, 97, 2990–3011. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, L.; Guo, X.; Wang, Z.; Lian, W.; Deng, K.; Lu, L.; Xing, B.; Zhu, H. Pituitary adenomas in patients with multiple endocrine neoplasia type 1: A single-center experience in China. Pituitary 2019, 22, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Gadelha, M.R.; Kasuki, L.; Korbonits, M. The genetic background of acromegaly. Pituitary 2017, 20, 10–21. [Google Scholar] [CrossRef]
- de Laat, J.M.; Dekkers, O.M.; Pieterman, C.R.; Kluijfhout, W.P.; Hermus, A.R.; Pereira, A.M.; van der Horst-Schrivers, A.N.; Drent, M.L.; Bisschop, P.H.; Havekes, B.; et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J. Clin. Endocrinol. Metab. 2015, 100, 3288–3296. [Google Scholar] [CrossRef]
- Pieterman, C.R.C.; Waguespack, S.G. Chapter 9—Multiple endocrine neoplasia syndromes and somatotroph adenomas. In Gigantism and Acromegaly; Stratakis, C.A., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 173–195. [Google Scholar] [CrossRef]
- Chevalier, B.; Coppin, L.; Romanet, P.; Cuny, T.; Maiza, J.C.; Abeillon, J.; Forestier, J.; Walter, T.; Gilly, O.; Le Bras, M.; et al. Beyond MEN1, when to think about MEN4? Retrospective study on 5600 patients in the French population & literature review. J. Clin. Endocrinol. Metab. 2024, 109, e1482–e1493. [Google Scholar] [CrossRef] [PubMed]
- Singeisen, H.; Renzulli, M.M.; Pavlicek, V.; Probst, P.; Hauswirth, F.; Muller, M.K.; Adamczyk, M.; Weber, A.; Kaderli, R.M.; Renzulli, P. Multiple endocrine neoplasia type 4: A new member of the MEN family. Endocr. Connect. 2023, 12, e220411. [Google Scholar] [CrossRef] [PubMed]
- Halperin, R.; Arnon, L.; Nasirov, S.; Friedensohn, L.; Gershinsky, M.; Telerman, A.; Friedman, E.; Bernstein-Molho, R.; Tirosh, A. Germline CDKN1B variant type and site are associated with phenotype in MEN4. Endocr. Relat. Cancer 2023, 30, e220174. [Google Scholar] [CrossRef]
- Tatsi, C.; Pitsava, G.; Faucz, F.R.; Keil, M.; Stratakis, C.A. The spectrum of growth hormone excess in Carney complex and genotype-phenotype correlations. J. Clin. Endocrinol. Metab. 2024, dgae253. [Google Scholar] [CrossRef] [PubMed]
- Groussin, L.; Horvath, A.; Jullian, E.; Boikos, S.; Rene-Corail, F.; Lefebvre, H.; Cephise-Velayoudom, F.L.; Vantyghem, M.C.; Chanson, P.; Conte-Devolx, B.; et al. A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J. Clin. Endocrinol. Metab. 2006, 91, 1943–1949. [Google Scholar] [CrossRef]
- Forlino, A.; Vetro, A.; Garavelli, L.; Ciccone, R.; London, E.; Stratakis, C.A.; Zuffardi, O. PRKACB and Carney complex. N. Engl. J. Med. 2014, 370, 1065–1067. [Google Scholar] [CrossRef]
- Salenave, S.; Boyce, A.M.; Collins, M.T.; Chanson, P. Acromegaly and McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 2014, 99, 1955–1969. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright syndrome. Orphanet J. Rare Dis. 2008, 3, 12. [Google Scholar] [CrossRef]
- Lecumberri, B.; Pozo-Kreilinger, J.J.; Esteban, I.; Gomes, M.; Royo, A.; Gomez de la Riva, A.; Perez de Nanclares, G. Head and neck manifestations of an undiagnosed McCune-Albright syndrome: Clinicopathological description and literature review. Virchows Arch. 2018, 473, 645–648. [Google Scholar] [CrossRef]
- Jayant, S.S.; Walia, R.; Gupta, R.; Pal, R.; Chaudhary, S.; Agrawal, K.; Rastogi, A.; Bhattacharya, A.; Dutta, P.; Bhadada, S.K.; et al. Autonomous growth hormone secretion due to McCune Albright syndrome in paediatric age group: An ominous triad. Endocrine 2023, 81, 149–159. [Google Scholar] [CrossRef] [PubMed]
- NIH. NIH: National Library of Medicine; Gene ID: 2778. Available online: https://www.ncbi.nlm.nih.gov/gene/2778 (accessed on 17 April 2024).
- Balinisteanu, I.; Panzaru, M.C.; Caba, L.; Ungureanu, M.C.; Florea, A.; Grigore, A.M.; Gorduza, E.V. Cancer Predisposition Syndromes and Thyroid Cancer: Keys for a Short Two-Way Street. Biomedicines 2023, 11, 2143. [Google Scholar] [CrossRef]
- Vado, Y.; Manero-Azua, A.; Pereda, A.; Perez de Nanclares, G. Choosing the Best Tissue and Technique to Detect Mosaicism in Fibrous Dysplasia/McCune-Albright Syndrome (FD/MAS). Genes 2024, 15, 120. [Google Scholar] [CrossRef]
- Roszko, K.L.; Guthrie, L.; Li, X.; Collins, M.T.; de Castro, L.F.; Boyce, A.M. Identification of GNAS Variants in Circulating Cell-Free DNA from Patients with Fibrous Dysplasia/McCune Albright Syndrome. J. Bone Min. Res. 2023, 38, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Stutz, B.; Korbonits, M.; Kothbauer, K.; Muller, W.; Fischli, S. Identification of a TMEM127 variant in a patient with paraganglioma and acromegaly. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Seabrook, A.J.; Harris, J.E.; Velosa, S.B.; Kim, E.; McInerney-Leo, A.M.; Dwight, T.; Hockings, J.I.; Hockings, N.G.; Kirk, J.; Leo, P.J.; et al. Multiple Endocrine Tumors Associated with Germline MAX Mutations: Multiple Endocrine Neoplasia Type 5? J. Clin. Endocrinol. Metab. 2021, 106, 1163–1182. [Google Scholar] [CrossRef] [PubMed]
- Mamedova, E.; Vasilyev, E.; Petrov, V.; Buryakina, S.; Tiulpakov, A.; Belaya, Z. Familial Acromegaly and Bilateral Asynchronous Pheochromocytomas in a Female Patient With a MAX Mutation: A Case Report. Front. Endocrinol. 2021, 12, 683492. [Google Scholar] [CrossRef] [PubMed]
- Denes, J.; Swords, F.; Rattenberry, E.; Stals, K.; Owens, M.; Cranston, T.; Xekouki, P.; Moran, L.; Kumar, A.; Wassif, C.; et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: Results from a large patient cohort. J. Clin. Endocrinol. Metab. 2015, 100, E531–E541. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, S.M.; Denes, J.; Robledo, M.; Stratakis, C.A.; Korbonits, M. 15 YEARS OF PARAGANGLIOMA: The association of pituitary adenomas and phaeochromocytomas or paragangliomas. Endocr. Relat. Cancer 2015, 22, T105–T122. [Google Scholar] [CrossRef] [PubMed]
- Hozumi, K.; Fukuoka, H.; Odake, Y.; Takeuchi, T.; Uehara, T.; Sato, T.; Inoshita, N.; Yoshida, K.; Matsumoto, R.; Bando, H.; et al. Acromegaly caused by a somatotroph adenoma in patient with neurofibromatosis type 1. Endocr. J. 2019, 66, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Man, A.; Di Scipio, M.; Grewal, S.; Suk, Y.; Trinari, E.; Ejaz, R.; Whitney, R. The Genetics of Tuberous Sclerosis Complex and Related mTORopathies: Current Understanding and Future Directions. Genes 2024, 15, 332. [Google Scholar] [CrossRef]
- Northrup, H.; Krueger, D.A.; International Tuberous Sclerosis Complex Consensus, G. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol. 2013, 49, 243–254. [Google Scholar] [CrossRef]
- Wang, M.X.; Segaran, N.; Bhalla, S.; Pickhardt, P.J.; Lubner, M.G.; Katabathina, V.S.; Ganeshan, D. Tuberous Sclerosis: Current Update. Radiographics 2021, 41, 1992–2010. [Google Scholar] [CrossRef]
- Hernandez-Ramirez, L.C.; Perez-Rivas, L.G.; Theodoropoulou, M.; Korbonits, M. An Update on the Genetic Drivers of Corticotroph Tumorigenesis. Exp. Clin. Endocrinol. Diabetes 2024. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Sheriff, M.O.; Garg, B.R.; Ratnakar, C. A rare association of localized gigantism with tuberous sclerosis. J. Dermatol. 1992, 19, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Ortonne, J.P.; Jeune, R.; Fulton, R.; Thivolet, J. Primary localized gigantism and tuberous sclerosis. Arch. Dermatol. 1982, 118, 877–878. [Google Scholar] [CrossRef]
- Colamaria, V.; Zambelli, L.; Tinazzi, P.; Dalla Bernardina, B. Tuberous sclerosis associated with partial gigantism in a child. Brain Dev. 1988, 10, 178–181. [Google Scholar] [CrossRef]
- Prigent, F.; Foldes, C.; Laval-Jeantet, M.; Mabrouki, F.; Civatte, J. Bourneville’s tuberous sclerosis and monomelic gigantism. Ann. Dermatol. Venereol. 1983, 110, 755–756. [Google Scholar] [PubMed]
- Bhatkar, S.R.; Takkar, A. Focal gigantism in tuberous sclerosis. Int. J. Epilepsy 2017, 4, 104–105. [Google Scholar] [CrossRef]
- Brandi, M.L.; Gagel, R.F.; Angeli, A.; Bilezikian, J.P.; Beck-Peccoz, P.; Bordi, C.; Conte-Devolx, B.; Falchetti, A.; Gheri, R.G.; Libroia, A.; et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J. Clin. Endocrinol. Metab. 2001, 86, 5658–5671. [Google Scholar] [CrossRef] [PubMed]
- Chenlo, M.; Rodriguez-Gomez, I.A.; Serramito, R.; Garcia-Rendueles, A.R.; Villar-Taibo, R.; Fernandez-Rodriguez, E.; Perez-Romero, S.; Suarez-Farina, M.; Garcia-Allut, A.; Cabezas-Agricola, J.M.; et al. Unmasking a new prognostic marker and therapeutic target from the GDNF-RET/PIT1/p14ARF/p53 pathway in acromegaly. EBioMedicine 2019, 43, 537–552. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Type | Disease/Syndrome | Gene(s) | Location | Inheritance | Penetrance | Prevalence of Acromegaly | Clinical Particularities | Histological Particularities | Treatment Particularities | References |
|---|---|---|---|---|---|---|---|---|---|---|
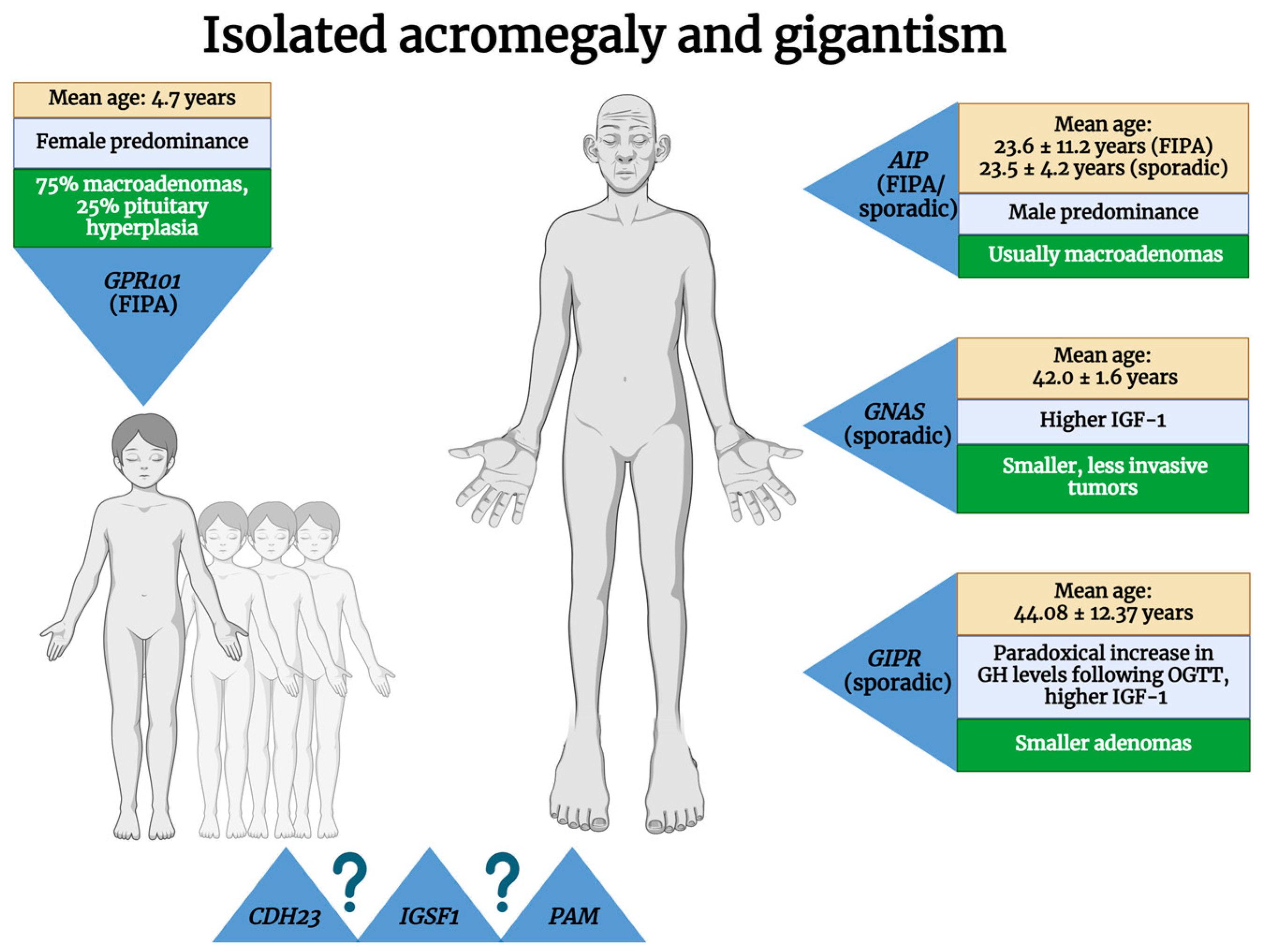
| Isolated | FIPAs | AIP (germline) | 11q13.2 | AD | 20–33% | 10–15% of FIPA families | Male predominance; age at diagnosis: median 23 years, mean 23.6 ± 11.2 years | Usually macroadenomas, sparsely granulated, low SSR2 expression | Resistance to I gen SAs; need for radiotherapy, repeated surgery, and multimodal therapy more often | [11,14,15,16,17,18,19,20,21,22,23] |
| GPR101 (germline + somatic) | Xq26.3 | X-linked | 100%, but 72.2% are de novo mutations | 7.8% of FIPAs | Female predominance; earlier age at diagnosis (median, 3.4–4.4 years; mean, 4.7 years) | 75% macroadenomas; 25% pituitary hyperplasia; generally, SSR2 is present, but SSR5 is variably present | SAs show poor results, Pegvisomant is more effective | [24,25,26,27] | ||
| CDH23 | 10q22.1 | ? | ? | Proposed as FIPA members, possible generators of GH-secreting PitNETs | ? | ? | ? | [22,28,29,30] | ||
| IGSF1 | Xq26.1 | |||||||||
| PAM | 5q21.1 | |||||||||
| Sporadic defects in genes involved in other diseases/syndromes | AIP (germline) | 11q13.2 | ? | ? | 3.4–5% of AIP-positive cases | Male predominance; age at diagnosis: mean, 23.5 ± 4.2 years | Usually macroadenomas, sparsely granulated, low SSR2 expression | 8% of SA-resistant cases | [14,19,31,32,33] | |
| GNAS (somatic) | 20q13.32 | ? | ? | 30–40% of GH-secreting PitNETs | Older patients (age at diagnosis: mean, 42.0 ± 1.6 years; median, 41 years), higher IGF-1 | Smaller, less invasive tumors; usually densely granulated; higher levels of DRD2 | Improved responses to surgery and to I gen SAs | [34,35,36,37,38,39,40,41,42,43] | ||
| Sporadic higher GIPR expression | GIPR | 19q13.32 | ? | ? | 24–27.4% of acromegaly | Paradoxical increase in GH levels following OGTT; higher IGF-1; mean age at diagnosis: 44.08 ± 12.37 years | Smaller densely granulated adenomas; ectopic expression of GIP receptor in somatotropinomas | Good response to SAs | [22,44,45,46] | |
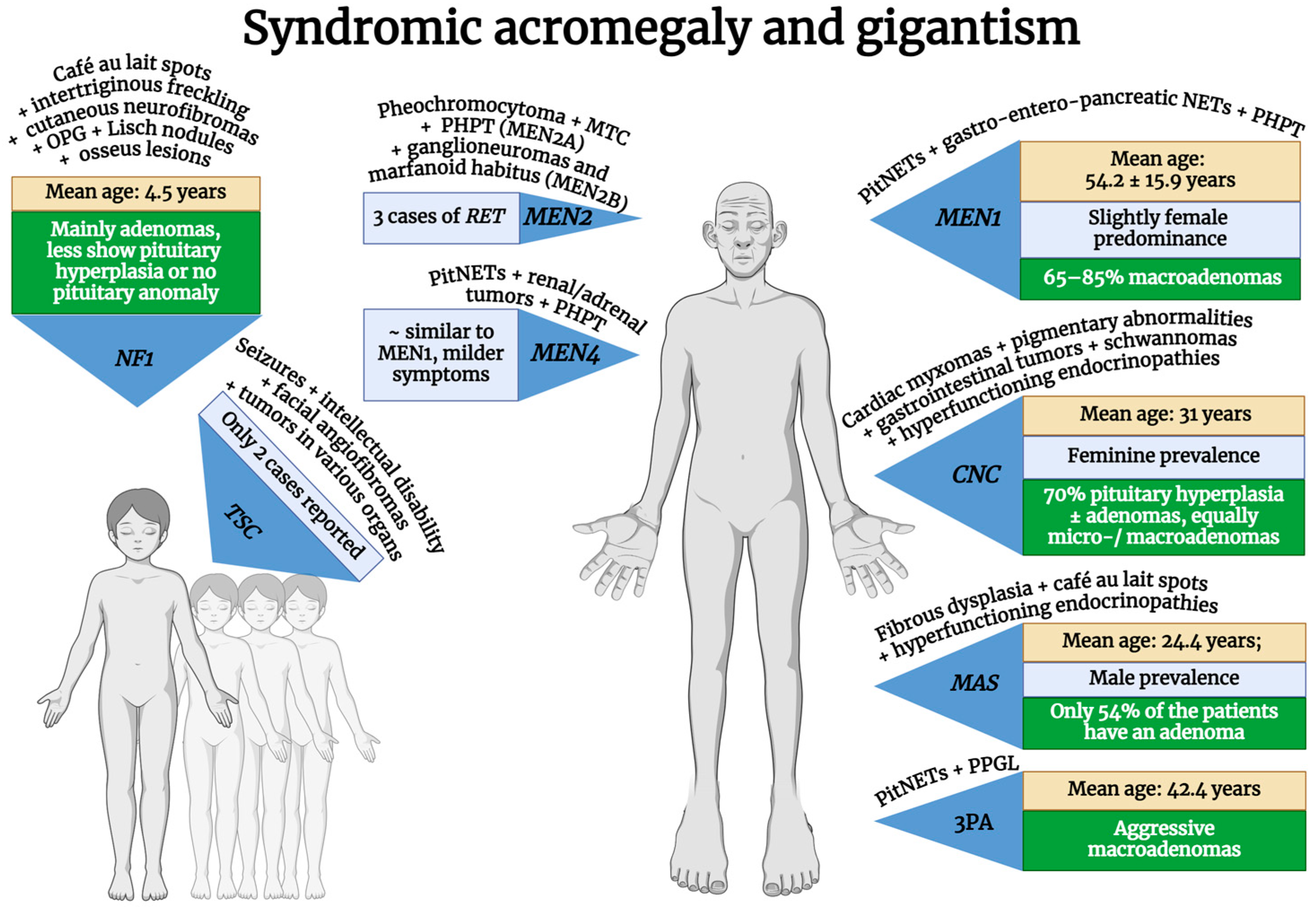
| Syndromic | MEN1 | MEN1 (mostly germline, but somatic variants exist too) | 11q13.1 | AD | 100% | 2.9–18.5% of acromegaly | Slight female predominance (59.1%); older age (mean 54.2 ± 15.9 years) | 65–85% macroadenomas | Poor response to drug therapy but better prognosis compared to AIP-positive adenomas | [10,47,48,49,50,51,52,53,54,55,56,57] |
| MEN2 | RET (germline, but in some cancers can be somatic) | 10q11.21 | AD | 100% | Three cases of RET mutations reported | 44 (48-year-old female; 35-year-old male) | Prolactin co-secretion in two cases (data available only for two cases) | ? | [22,58,59,60,61] | |
| MEN4 | CDKN1B (germline) | 12p13.1 | AD | Most probably 100% | 7–10% of MEN4 | Similar to MEN1 but milder symptoms and undetermined mean age | ? | Standard therapy | [3,22,40,62,63,64,65] | |
| Carney complex type 1 | PRKAR1A | 17q24.2 | 70% AD, 30% de novo | >95% | 10% of the CNC | Feminine prevalence. Age at diagnosis: median, 25.3–28.8 years; mean, 31 years | 70% pituitary hyperplasia ± adenomas; same proportion of micro- and macroadenomas | Probably normal response: a small proportion of subjects are considered resistant to treatment | [10,22,54,66,67,68,69,70,71,72] | |
| PDE11A | 2q31.2 | |||||||||
| PRKACB | 1p31.1 | |||||||||
| PRKACA | 19p13.12 | |||||||||
| (germline) | ||||||||||
| McCune–Albright syndrome | GNAS (somatic–mosaic) | 20q13.32 | Not inherited | - | 20–30% of MAS | Male prevalence: mean age at diagnosis is 24.4 years | Only 54% of the patients have an adenoma | Poor response to surgery alone; 70% are not fully controlled under I gen SAs; Pegvisomant and Pasireotide have good response | [73,74,75,76] | |
| 3P association | MAX | 14q23.3 | AD | ? | Extremely rare | Higher age of onset than AIP/MEN1: mean, 42.4 years | Aggressive macroadenomas, intracytoplasmic vacuoles | Patients with MAX mutations require a multimodal treatment approach | [3,10,77,78,79] | |
| SDHA | 5p15.33 | |||||||||
| SDHAF2 | 11q12.2 | |||||||||
| SDHB | 1p36.13 | |||||||||
| SDHC | 1q23.3 | |||||||||
| SDHD | 11q23.1 | |||||||||
| TMEM127 | 2q11.2 | |||||||||
| (germline) | ||||||||||
| Neurofibromatosis type 1 | NF1 (germline, but in some cancers can be somatic) | 17q11.2 | AD | 100% | 10% of NF1 | Mainly seen in children but can also appear in adolescents and adults; mean age at diagnosis: 4.5 years | Most are pituitary adenomas, some are pituitary hyperplasia, and some have no pituitary anomalies at all; a significant proportion of patients also have OPG | In NF1 children with OPG, GH excess can be reversed, and short-term SA therapy could be sufficient | [10,80,81,82,83,84,85,86] | |
| Tuberous sclerosis complex | TSC1 | 9q34.13 | AD, though expressivity is variable | 100% | Only two cases reported | Age at diagnosis: 9 years old (available for just one case) | Macroadenoma (available for just one case); Prolactin co-secretion (two cases) | ? | [22,87,88,89] | |
| TSC2 | 16p13.3 | |||||||||
| (germline) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balinisteanu, I.; Caba, L.; Florea, A.; Popescu, R.; Florea, L.; Ungureanu, M.-C.; Leustean, L.; Gorduza, E.V.; Preda, C. Unlocking the Genetic Secrets of Acromegaly: Exploring the Role of Genetics in a Rare Disorder. Curr. Issues Mol. Biol. 2024, 46, 9093-9121. https://doi.org/10.3390/cimb46080538
Balinisteanu I, Caba L, Florea A, Popescu R, Florea L, Ungureanu M-C, Leustean L, Gorduza EV, Preda C. Unlocking the Genetic Secrets of Acromegaly: Exploring the Role of Genetics in a Rare Disorder. Current Issues in Molecular Biology. 2024; 46(8):9093-9121. https://doi.org/10.3390/cimb46080538
Chicago/Turabian StyleBalinisteanu, Ioana, Lavinia Caba, Andreea Florea, Roxana Popescu, Laura Florea, Maria-Christina Ungureanu, Letitia Leustean, Eusebiu Vlad Gorduza, and Cristina Preda. 2024. "Unlocking the Genetic Secrets of Acromegaly: Exploring the Role of Genetics in a Rare Disorder" Current Issues in Molecular Biology 46, no. 8: 9093-9121. https://doi.org/10.3390/cimb46080538
APA StyleBalinisteanu, I., Caba, L., Florea, A., Popescu, R., Florea, L., Ungureanu, M.-C., Leustean, L., Gorduza, E. V., & Preda, C. (2024). Unlocking the Genetic Secrets of Acromegaly: Exploring the Role of Genetics in a Rare Disorder. Current Issues in Molecular Biology, 46(8), 9093-9121. https://doi.org/10.3390/cimb46080538