
The ER Stress Induced in Human Neuroblastoma Cells Can Be Reverted by Lumacaftor, a CFTR Corrector

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
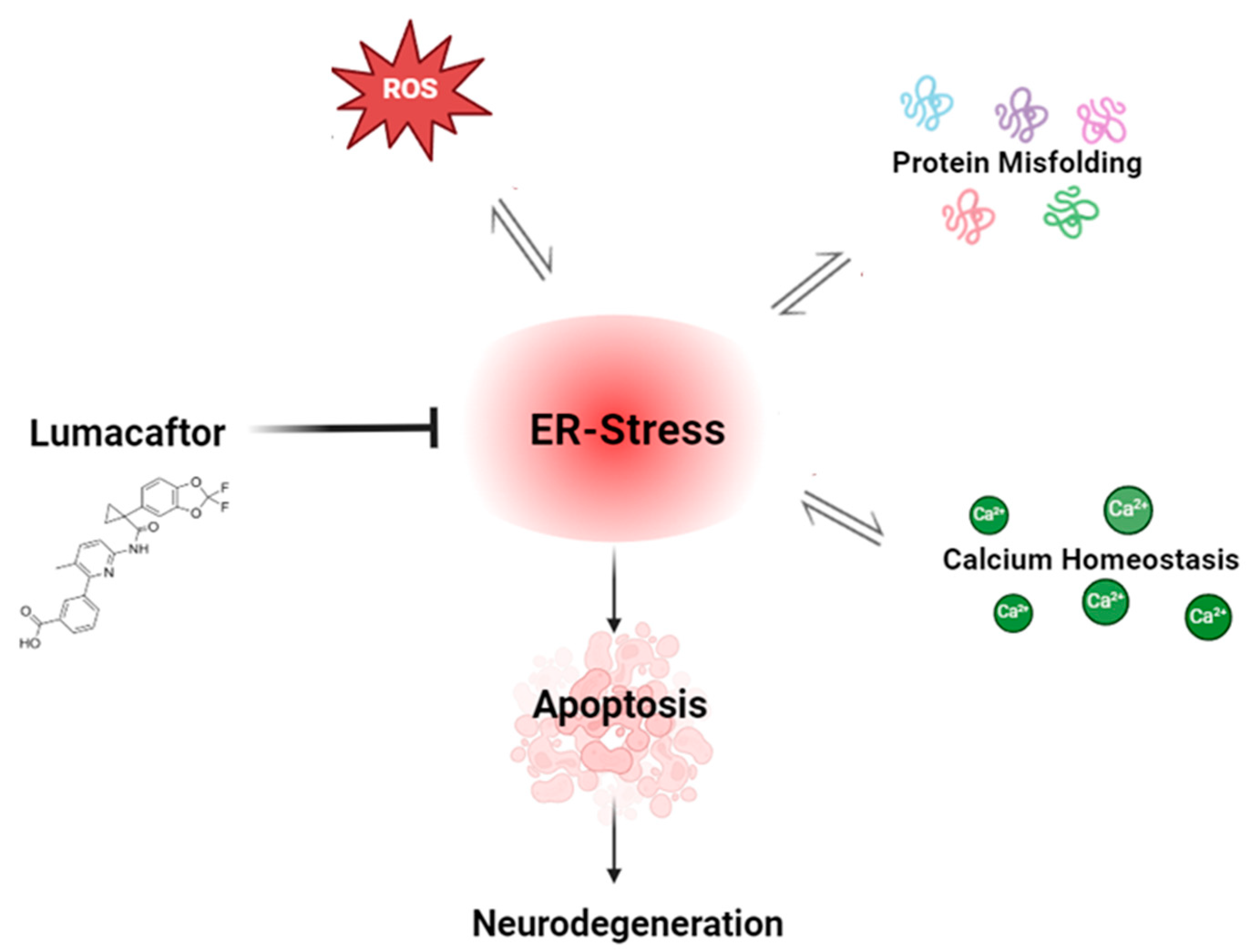
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Experimental Protocol
2.4. Cell Viability
2.5. Protein Extraction and Western Blotting Assay
2.6. RNA Extraction and Real-Time RT-PCR Protocol
2.7. Calcium Signaling Assay
2.8. Intracellular and Mitochondrial ROS Detection
2.9. Caspase 4 Measurement
2.10. Hypodiploid DNA Detection
2.11. Analytical Statistics
3. Results
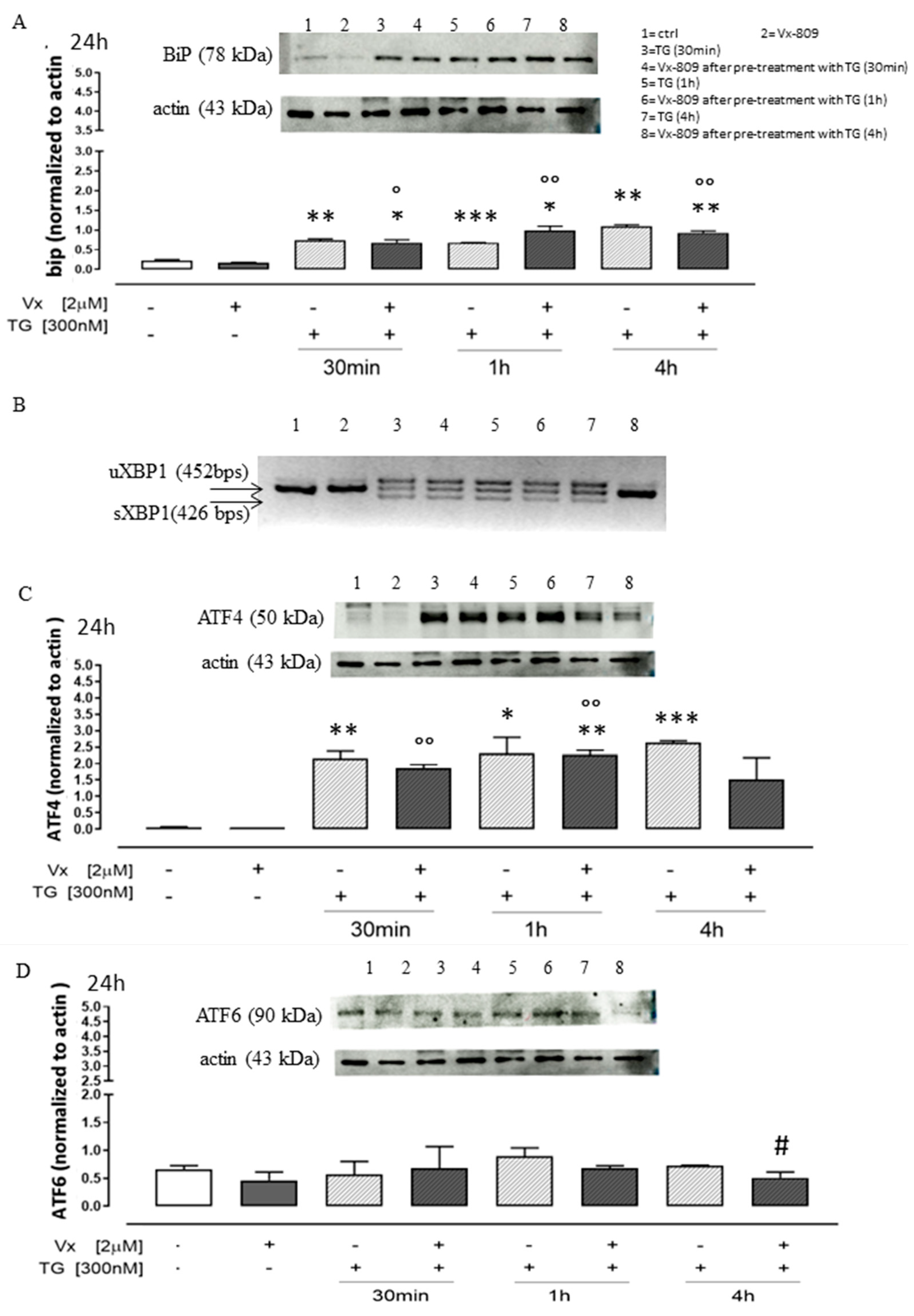
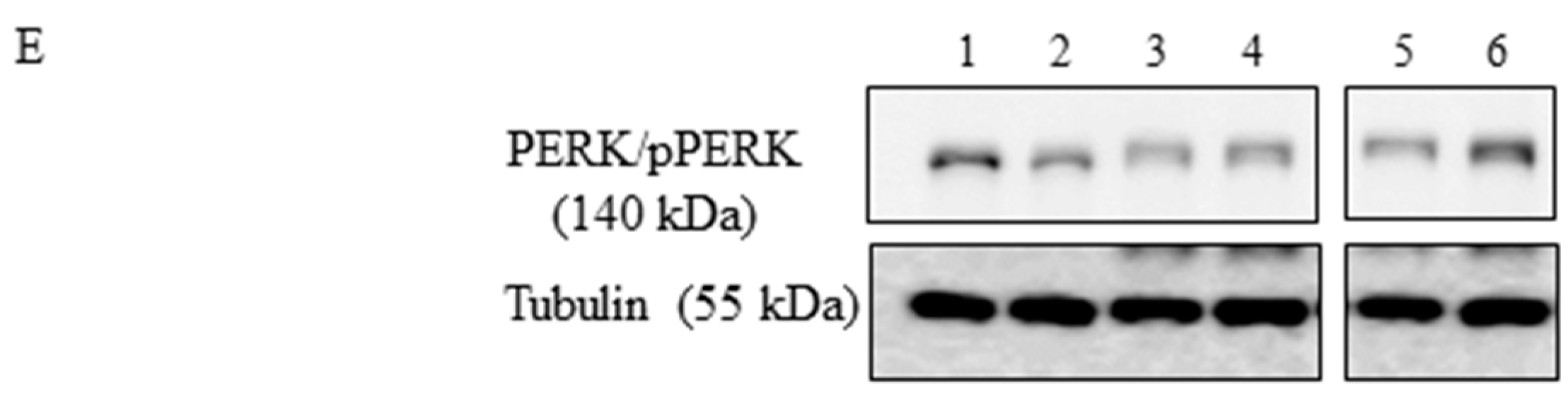
3.1. Role of Vx-809 in UPR Pathway
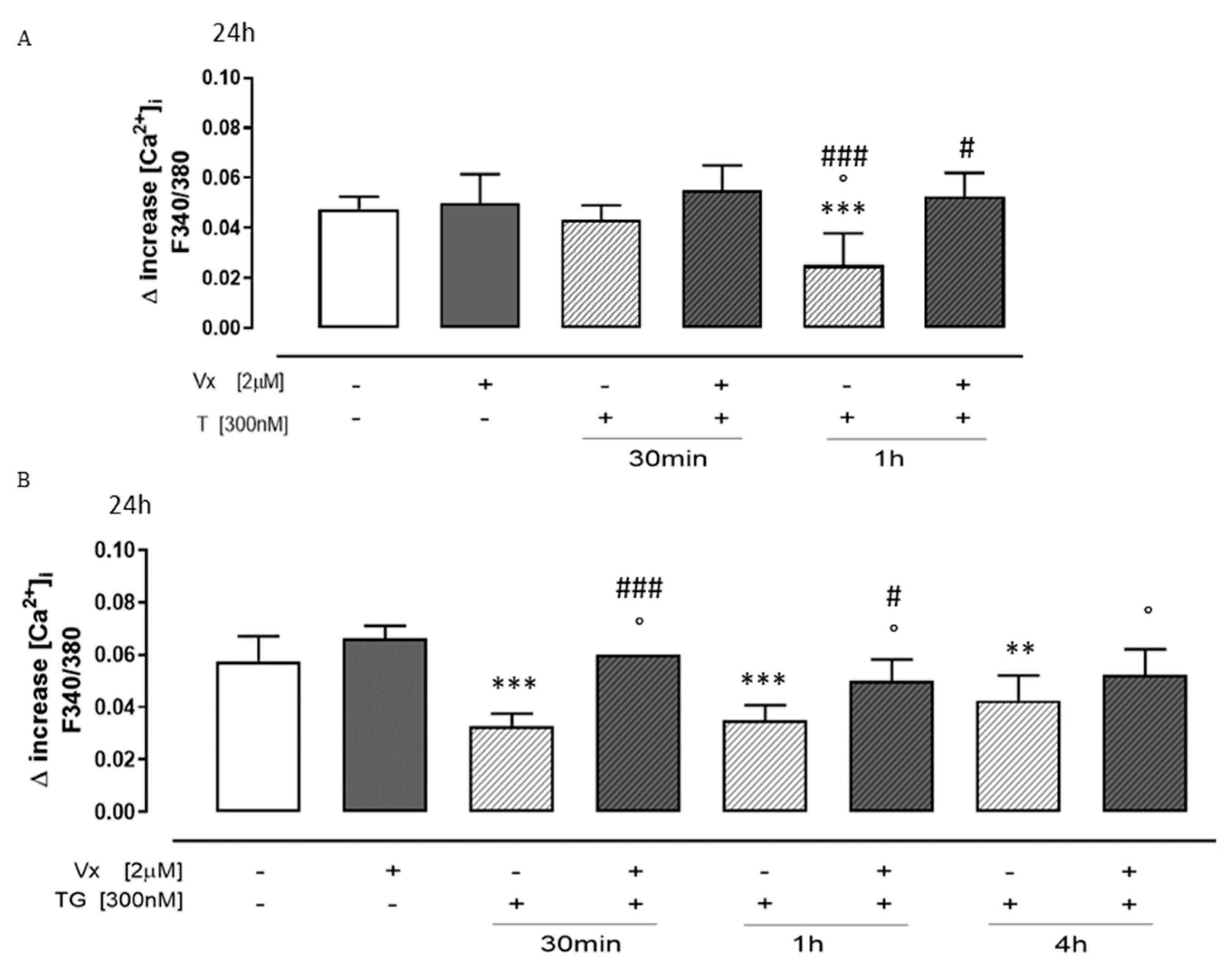
3.2. “Corrector” Vx-809 Interferes on Calcium Signaling
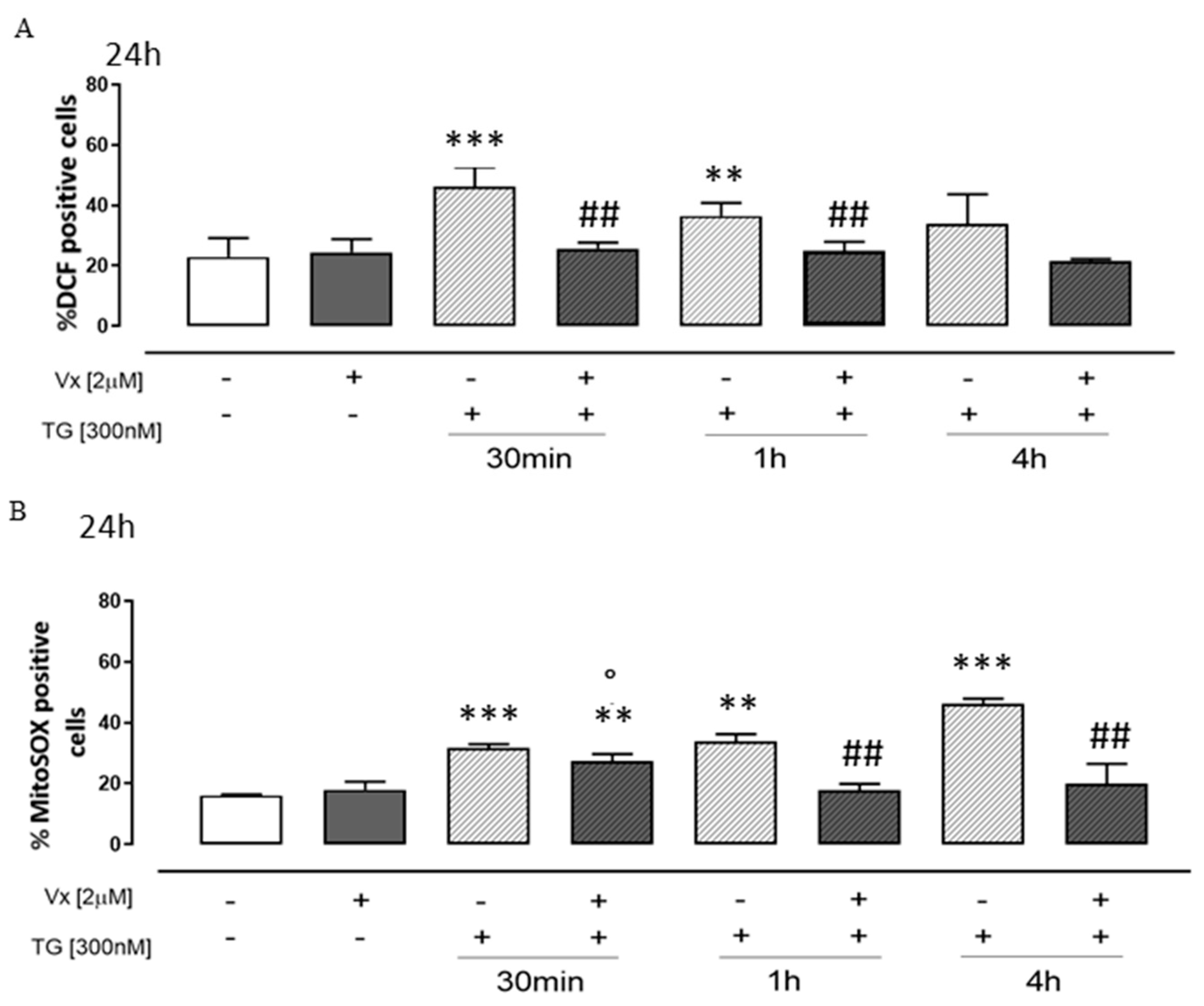
3.3. Vx-809 Counteract Thapsigargin-Induced Oxidative Stress
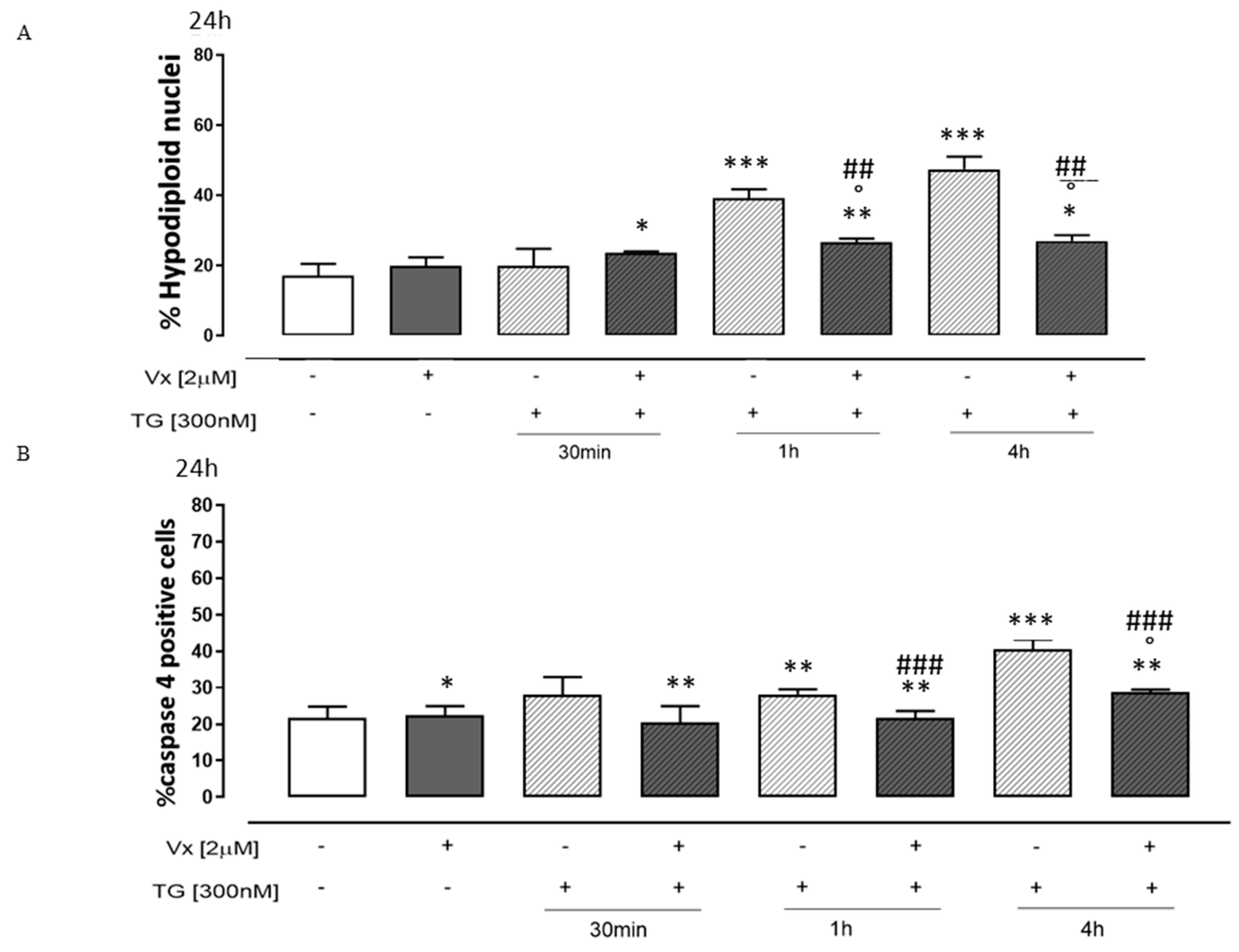
3.4. Vx-809 Interferes with the Apoptotic Pathway
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ER | Endoplasmic reticulum |
| UPR | Unfolded protein response |
| IRE1α | Inositol-requiring enzyme 1 α |
| PERK | Protein kinase RNA-like ER kinase |
| ATF6 | Activating transcription Factor 6 |
| GRP78/BiP | Glucose-regulated protein 78/binding immunoglobulin protein |
| eIF2α | Eukaryotic translation initiation factor 2A |
| ATF4 | Activating transcription factor 4 |
| CHOP | C/EBP homologous protein |
| ERAD | ER-associated degradation |
| Vx-809 | Lumacaftor |
| CF | Cystic fibrosis |
| NBD1 | Nucleotide-binding domain 1 |
| TMD1 | Transmembrane domain 1 |
| TG | Thapsygargin |
| NDs | Neurodegenerative diseases |
| ROS | Reactive oxygen species |
| PD | Parkinson’s disease |
| PrD | Prion disease |
| Xbp1 | X-box binding protein 1 |
| Xbp1s | Spliced Xbp1 |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| SODIII | Superoxide dismutase III |
| DMEM | Dulbecco’s modified Eagle’s Medium |
| FBS | Fetal bovine serum |
| PBS | Phosphate-buffered saline |
| MTT | 3-[4,5-dimetiltiazol]-2,5-diphenyl-2H-tetrazolium bromide |
| Sp | Staurosporine |
| ECL | Enhanced chemiluminescence |
| HBSS | Hank’s balanced salt solution |
| H2DCF-DA | 2′,7′-dichlorofluorescin diacetate |
| MitoSOX | Mitochondrial superoxide |
| PI | Propidium iodide |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| ALSIDP | Amyotrophic lateral sclerosisIntrinsically disordered proteins |
References
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, B.; Berlow, H.; Dyson, J.; Peter, E. Wright, Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J. Mol. Biol. 2018, 430, 2309–2320. [Google Scholar]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Ashraf, G.; Greig, N.; Khan, T.; Hassan, I.; Tabrez, S.; Shakil, S.; Sheikh, I.; Zaidi, S.; Akram, M.; Jabir, N.; et al. Protein Misfolding and Aggregation in Alzheimer’s Disease and Type 2 Diabetes Mellitus. CNS Neurol. Disord. Drug Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Frakes, A.E.; Dillin, A. The UPRER: Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 2017, 66, 761–771. [Google Scholar] [CrossRef]
- Li, Y.; Gao, S.; Meng, Y. Integrated analysis of endoplasmic reticulum stress regulators’ expression identifies distinct subtypes of autism spectrum disorder. Front. Psychiatry 2023, 17, 1136154. [Google Scholar] [CrossRef]
- Pecoraro, M.; Serra, A.; Pascale, M.; Franceschelli, S. Vx-809, a CFTR Corrector, Acts through a General Mechanism of Protein Folding and on the Inflammatory Process. Int. J. Mol. Sci. 2023, 24, 4252. [Google Scholar] [CrossRef]
- Freeman, O.J.; Mallucci, G.R. The UPR and synaptic dysfunction in neurodegeneration. Brain Res. 2016, 1648, 530–537. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Lozada Ortiz, J.; Betancor, M.; Pérez Lázaro, S.; Bolea, R.; Badiola, J.J.; Otero, A. Endoplasmic reticulum stress and ubiquitin-proteasome system impairment in natural scrapie. Front. Mol. Neurosci. 2023, 16, 1175364. [Google Scholar] [CrossRef] [PubMed]
- Kabir, F.; Atkinson, R.; Cook, A.L.; Phipps, A.J.; King, A.E. The role of altered protein acetylation in neurodegenerative disease. Front. Aging Neurosci. 2023, 14, 1025473. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef]
- Angelova, P.R.; Ludtmann, M.H.R.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ is a key factor in α-synuclein-induced neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef] [PubMed]
- Magrinelli, F.; Mehta, S.; Di Lazzaro, G.; Latorre, A.; Edwards, M.J.; Balint, B.; Basu, P.; Kobylecki, C.; Groppa, S.; Hegde, A.; et al. Dissecting the Phenotype and Genotype of PLA2G6-Related Parkinsonism. Mov. Disord. 2022, 37, 148–161. [Google Scholar] [CrossRef]
- Goldsteins, G.; Hakosalo, V.; Jaronen, M.; Keuters, M.H.; Lehtonen, Š.; Koistinaho, J. CNS Redox Homeostasis and Dysfunction in Neurodegenerative Diseases. Antioxidants 2022, 11, 405. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef]
- Pecoraro, M.; Franceschelli, S.; Pascale, M. Lumacaftor and Matrine: Possible Therapeutic Combination to Counteract the Inflammatory Process in Cystic Fibrosis. Biomolecules 2021, 11, 422. [Google Scholar] [CrossRef]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. Correctors of the Major Cystic Fibrosis Mutant Interact through Membrane-Spanning Domains. Mol. Pharmacol. 2018, 93, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Bardin, E.; Pastor, A.; Semeraro, M.; Golec, A.; Hayes, K.; Chevalier, B.; Berhal, F.; Prestat, G.; Hinzpeter, A.; Gravier-Pelletier, C.; et al. Modulators of CFTR. Updates on clinical development and future directions. Eur. J. Med. Chem. 2021, 213, 113195. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Pinto, A.; Popolo, A. Inhibition of Connexin 43 translocation on mitochondria accelerates CoCl2-induced apoptotic response in a chemical model of hypoxia. Toxicol. In Vitro 2018, 47, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Marzocco, S.; Belvedere, R.; Petrella, A.; Franceschelli, S.; Popolo, A. Simvastatin Reduces Doxorubicin-Induced Cardiotoxicity: Effects beyond Its Antioxidant Activity. Int. J. Mol. Sci. 2023, 24, 7573. [Google Scholar] [CrossRef]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Mai, K.; Ai, Q. Effects of GRP78 on Endoplasmic Reticulum Stress and Inflammatory Response in Macrophages of Large Yellow Croaker (Larimichthys crocea). Int. J. Mol. Sci. 2023, 24, 5855. [Google Scholar] [CrossRef]
- Yang, J.; Kim, K.S.; Iyirhiaro, G.O.; Marcogliese, P.C.; Callaghan, S.M.; Qu, D.; Kim, W.J.; Slack, R.S.; Park, D.S. DJ-1 modulates the unfolded protein response and cell death via upregulation of ATF4 following ER stress. Cell Death Dis. 2019, 10, 135. [Google Scholar] [CrossRef]
- Yu, Z.; Sheng, H.; Liu, S.; Zhao, S.L.; Glembotski, C.C.; Warneret, D.S.; Paschen, W.; Yang, W. Activation of the ATF6 branch of the unfolded protein response in neurons improves stroke outcome. J. Cereb. Blood Flow Metab. 2017, 37, 1069–1079. [Google Scholar] [CrossRef]
- Shen, Y.T.; Li, R.; Yu, S.; Zhao, Q.; Wang, Z.R.; Sheng, H.X.; Yang, W.V. Activation of the ATF6 (Activating Transcription Factor 6) signaling pathway in neurons improves outcome after cardiac arrest in mice. J. Am. Heart Assoc. 2021, 10, e020216. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Y.; Zhang, X.; Ye, Y.; Xiong, X.; Zhang, S.; Gu, L.; Jian, Z.; Wang, H. Endoplasmic Reticulum Stress and the Unfolded Protein Response in Cerebral Ischemia/Reperfusion Injury. Front. Cell. Neurosci. 2022, 16, 864426. [Google Scholar] [CrossRef]
- Mercado, G.; Castillo, V.; Soto, P.; Sidhu, A. ER stress and Parkinson’s disease: Pathological inputs that converge into the secretory pathway. Brain Res. 2016, 1648, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.K.; Jeong, S.; Lee, J. The Common Cellular Events in the Neurodegenerative Diseases and the Associated Role of Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2022, 23, 5894. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gao, N.; Hu, X.; Luo, H.; Peng, J.; Xia, Y. SOD3 overexpression alleviates cerebral ischemia-reperfusion injury in rats. Mol. Genet. Genom. Med. 2019, 7, e00831. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, Y.; Lu, Y.; Xiang, M. ROS and Endoplasmic Reticulum Stress in Pulmonary Disease. Front. Pharmacol. 2022, 13, 879204. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42 (Suppl. S3), S125–S152. [Google Scholar] [CrossRef]
- Chukai, Y.; Ito, G.; Konno, M.; Sakata, Y.; Ozaki, T. Mitochondrial calpain-5 truncates caspase-4 during endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2022, 608, 156–162. [Google Scholar] [CrossRef]
- Youssef, M.M.M.; Park, J. LONRF2 is a gatekeeper against protein aggregation in aging neurons. Nat. Aging 2023, 3, 913–914. [Google Scholar] [CrossRef] [PubMed]
- Candelise, N.; Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C.; Manganelli, V.; Garofalo, T.; Sorice, M.; Misasi, R. Protein Aggregation Landscape in Neurodegenerative Diseases: Clinical Relevance and Future Applications. Int. J. Mol. Sci. 2021, 22, 6016. [Google Scholar] [CrossRef] [PubMed]
- Coleman, O.I.; Haller, D. ER Stress and the UPR in Shaping Intestinal Tissue Homeostasis and Immunity. Front. Immunol. 2019, 10, 2825. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Castillo, K.; Armisén, R.; Stutzin, A.; Soto, C.; Hetz, C. Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS ONE 2010, 5, e15658. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER stress and UPR in Alzheimer’s disease: Mechanisms, pathogenesis, treatments. Cell Death Dis. 2022, 13, 706. [Google Scholar] [CrossRef]
- Regard, L.; Martin, C.; Burnet, E.; Da Silva, J.; Burgel, P.R. CFTR Modulators in People with Cystic Fibrosis: Real-World Evidence in France. Cells 2022, 11, 1769. [Google Scholar] [CrossRef]
- Peng, S.Y.; Tang, J.Y.; Lan, T.H.; Shiau, J.P.; Chen, K.L.; Jeng, J.H.; Yen, C.Y.; Chang, H.W. Oxidative-Stress-Mediated ER Stress Is Involved in Regulating Manoalide-Induced Antiproliferation in Oral Cancer Cells. Int. J. Mol. Sci. 2023, 24, 3987. [Google Scholar] [CrossRef]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef]
- Torres, M.; Encina, G.; Soto, C.; Hetz, C. Abnormal calcium homeostasis and protein folding stress at the ER: A common factor in familial and infectious prion disorders. Commun. Integr. Biol. 2011, 4, 258–261. [Google Scholar] [CrossRef]
- Esmaeili, Y.; Yarjanli, Z.; Pakniya, F.; Bidram, E.; Łos, M.J.; Eshraghi, M.; Klionsky, D.J.; Ghavami, S.; Zarrabi, A. Targeting autophagy, oxidative stress, and ER stress for neurodegenerative disease treatment. J. Control Release 2022, 345, 147–175. [Google Scholar] [CrossRef]
- Karvandi, M.S.; Sheikhzadeh Hesari, F.; Aref, A.R.; Mahdavi, M. The neuroprotective effects of targeting key factors of neuronal cell death in neurodegenerative diseases: The role of ER stress, oxidative stress, and neuroinflammation. Front. Cell. Neurosci. 2023, 17, 1105247. [Google Scholar] [CrossRef]
- Ochneva, A.; Zorkina, Y.; Abramova, O.; Pavlova, O.; Ushakova, V.; Morozova, A.; Zubkov, E.; Pavlov, K.; Gurina, O.; Chekhonin, V. Protein Misfolding and Aggregation in the Brain: Common Pathogenetic Pathways in Neurodegenerative and Mental Disorders. Int. J. Mol. Sci. 2022, 23, 14498. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecoraro, M.; Serra, A.; Pascale, M.; Franceschelli, S. The ER Stress Induced in Human Neuroblastoma Cells Can Be Reverted by Lumacaftor, a CFTR Corrector. Curr. Issues Mol. Biol. 2024, 46, 9342-9358. https://doi.org/10.3390/cimb46090553
Pecoraro M, Serra A, Pascale M, Franceschelli S. The ER Stress Induced in Human Neuroblastoma Cells Can Be Reverted by Lumacaftor, a CFTR Corrector. Current Issues in Molecular Biology. 2024; 46(9):9342-9358. https://doi.org/10.3390/cimb46090553
Chicago/Turabian StylePecoraro, Michela, Adele Serra, Maria Pascale, and Silvia Franceschelli. 2024. "The ER Stress Induced in Human Neuroblastoma Cells Can Be Reverted by Lumacaftor, a CFTR Corrector" Current Issues in Molecular Biology 46, no. 9: 9342-9358. https://doi.org/10.3390/cimb46090553
APA StylePecoraro, M., Serra, A., Pascale, M., & Franceschelli, S. (2024). The ER Stress Induced in Human Neuroblastoma Cells Can Be Reverted by Lumacaftor, a CFTR Corrector. Current Issues in Molecular Biology, 46(9), 9342-9358. https://doi.org/10.3390/cimb46090553