

A Theoretical Study of the Interaction of PARP-1 with Natural and Synthetic Inhibitors: Advances in the Therapy of Triple-Negative Breast Cancer
 , , and
, , and 
Abstract
:1. Introduction
2. Computational Details
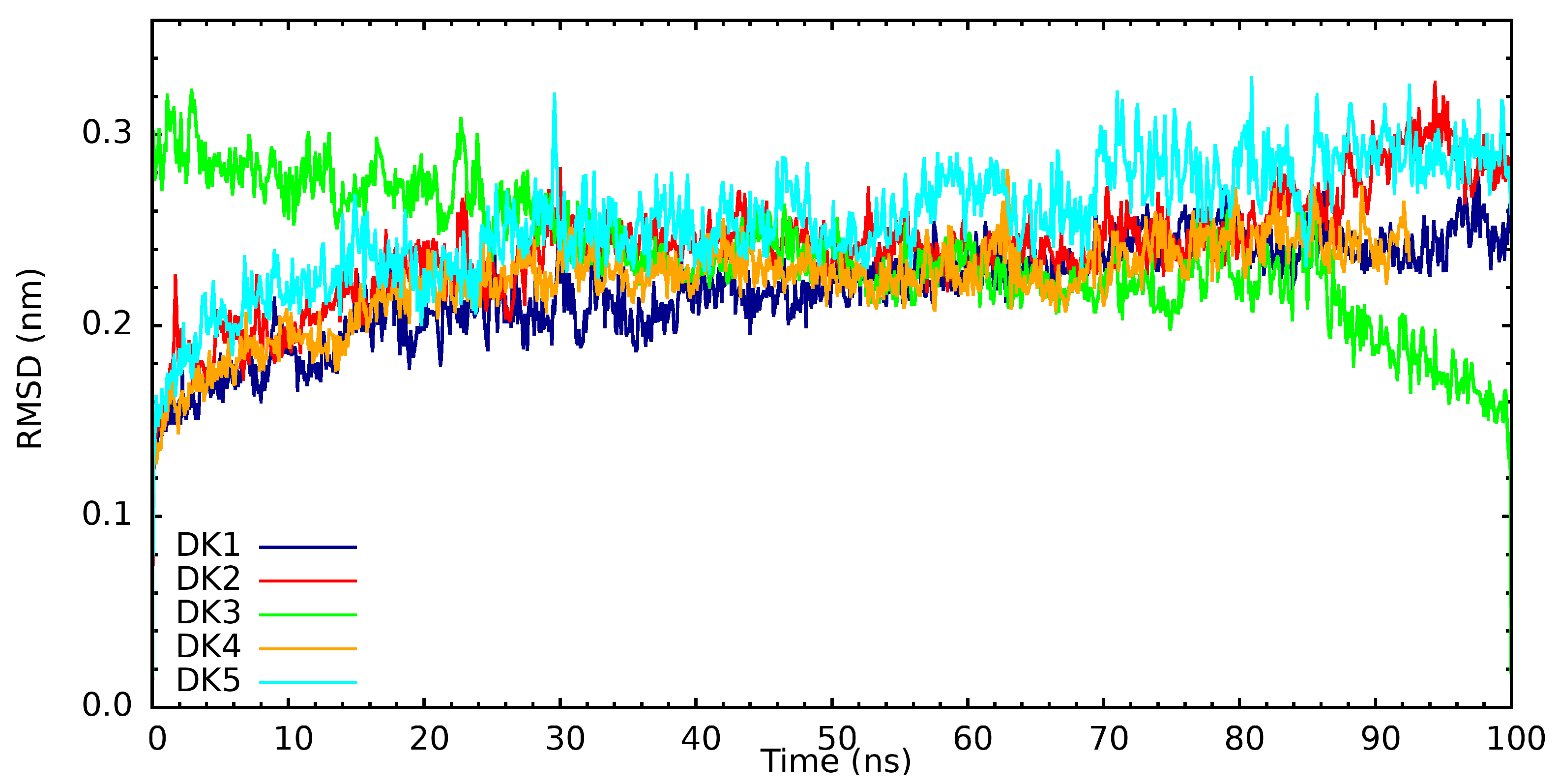
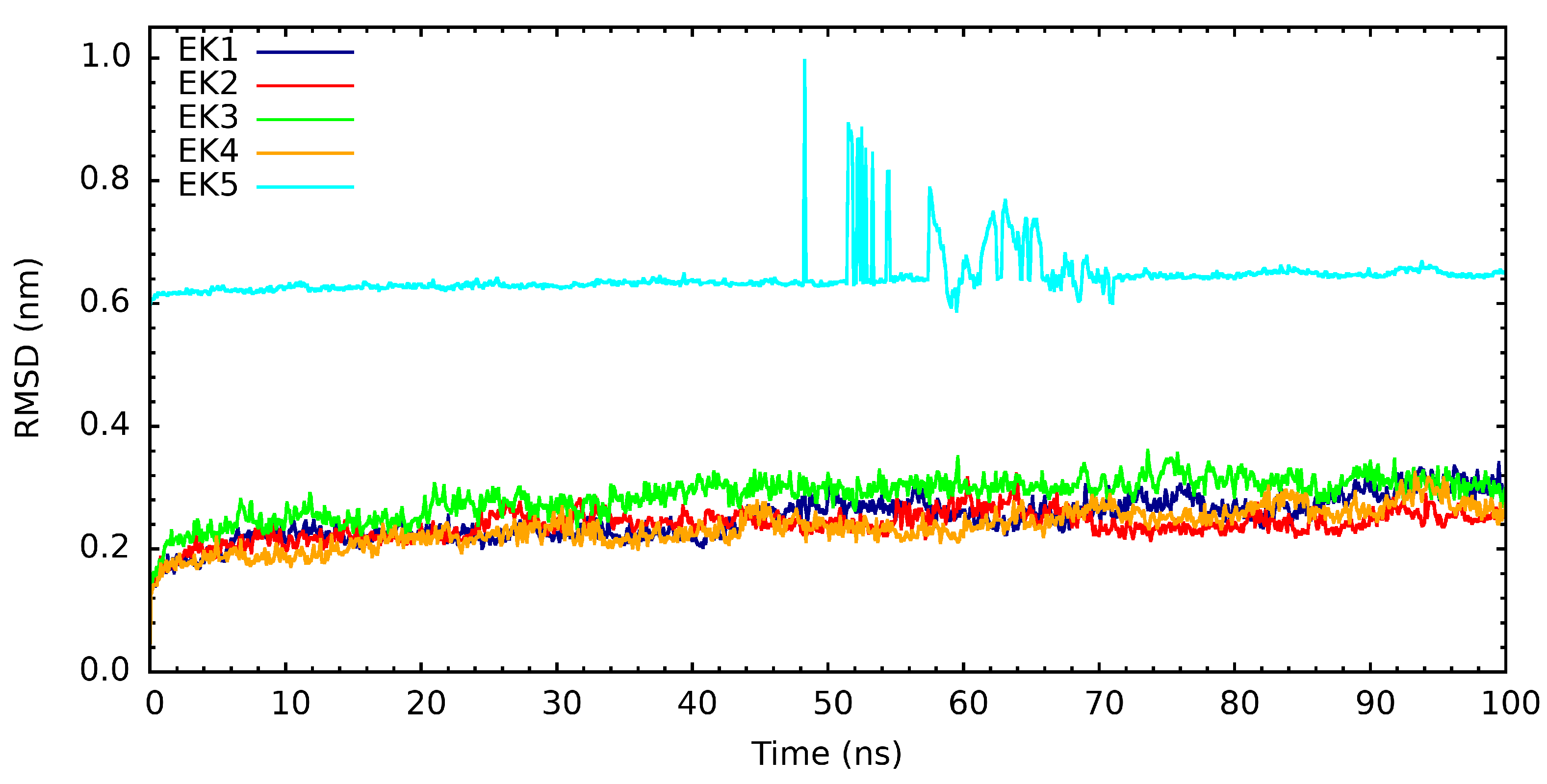
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramos Águila, Y.d.l.C.; Marimón Torres, E.R.; Crespo González, C.; Junco Sena, B.; Valiente Morejón, W. Cáncer de mama, su caracterización epidemiológica. Rev. De Cienc. Médicas De Pinar Del Río 2015, 19, 619–629. [Google Scholar]
- Vallejos-Sologuren, C.S. Situación del Cáncer en el Perú. Diagnóstico 2020, 59, 77–85. [Google Scholar] [CrossRef]
- Richard, I.A.; Burgess, J.T.; O’Byrne, K.J.; Bolderson, E. Beyond PARP1: The Potential of Other Members of the Poly(ADP-Ribose) Polymerase Family in DNA Repair and Cancer Therapeutics. Front. Cell Dev. Biol. 2022, 9, 801200. [Google Scholar] [CrossRef]
- Quiñonero Muñoz, F.J. Estudio de PARP-1 en Células Tumorales y Células Madre de Cáncer de Páncreas. Implicación en la Resistencia Tumoral y uso Como Diana Terapéutica. Ph.D. Thesis, Universidad de Granada, Granada, Spain, 2023. [Google Scholar]
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomarkers Amp; Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, B.; Nowak, A.Z.; Romanowicz, H. Breast Cancer—Epidemiology, Classification, Pathogenesis and Treatment (Review of Literature). Cancers 2022, 14, 2569. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, M.; Zeinomar, N.; Tehranifar, P.; Terry, M.B. Are Global Breast Cancer Incidence and Mortality Patterns Related to Country-Specific Economic Development and Prevention Strategies? J. Glob. Oncol. 2018, 4, 1–16. [Google Scholar] [CrossRef]
- Borri, F.; Granaglia, A. Pathology of triple negative breast cancer. Semin. Cancer Biol. 2021, 72, 136–145. [Google Scholar] [CrossRef]
- Gong, R.; Ma, Z.; He, L.; Jiang, S.; Cao, D.; Cheng, Y. Identification and evaluation of a novel PARP1 inhibitor for the treatment of triple-negative breast cancer. Chem. Biol. Interact. 2023, 382, 110567. [Google Scholar] [CrossRef]
- Rakha, E.A.; Ellis, I.O. Triple-negative/basal-like breast cancer: Review. Pathology 2009, 41, 40–47. [Google Scholar] [CrossRef]
- Mustacchi, G.; De Laurentiis, M. The role of taxanes in triple-negative breast cancer: Literature review. Drug Des. Devel. Ther. 2015, 9, 4303–4318. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, H.; Song, X.; Yang, Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin. Cancer Biol. 2020, 60, 14–27. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef]
- Feng, F.Y.; Speers, C.; Liu, M.; Jackson, W.C.; Moon, D.; Rinkinen, J.; Wilder-Romans, K.; Jagsi, R.; Pierce, L.J. Targeted radiosensitization with PARP1 inhibition: Optimization of therapy and identification of biomarkers of response in breast cancer. Breast Cancer Res. Treat. 2014, 147, 81–94. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- King, M.C.; Marks, J.H.; Mandell, J.B. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H.; Yang, D.; Yang, Q.; Tang, H.; Fu, Y.X.; Wan, Y. Differential regulation of breast cancer bone metastasis by PARP1 and PARP2. Nat. Commun. 2020, 11, 1578. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Lundine, D.; Annor, G.K.; Canar, J.; Ellison, V.; Polotskaia, A.; Donabedian, P.L.; Reiner, T.; Khramtsova, G.F.; Olopade, O.I.; et al. Gain-of-Function Mutant p53 R273H Interacts with Replicating DNA and PARP1 in Breast Cancer. Cancer Res. 2020, 80, 394–405. [Google Scholar] [CrossRef]
- Ohmoto, A.; Yachida, S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. Onco Targets Ther. 2017, 10, 5195–5208. [Google Scholar] [CrossRef] [PubMed]
- Lodovichi, S.; Mercatanti, A.; Cervelli, T.; Galli, A. Computational analysis of data from a genome-wide screening identifies new PARP1 functional interactors as potential therapeutic targets. Oncotarget 2019, 10, 2722–2737. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Chiarugi, A. A snapshot of chemoresistance to PARP inhibitors. Trends Pharmacol. Sci. 2012, 33, 42–48. [Google Scholar] [CrossRef]
- Montoni, A.; Robu, M.; Pouliot, E.; Shah, G.M. Resistance to PARP-Inhibitors in Cancer Therapy. Front. Pharmacol. 2013, 4, 18. [Google Scholar] [CrossRef]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Wei, Z.L.; Juan, W.; Tong, D.; Juan, L.X.; Sa, L.Y.; Jie, H.F.M.; Xiao, G.; Xiang, L.G.; Jie, H.M.; Xu, C. Curcumol inhibits breast cancer growth via NCL/ERα36 and the PI3K/AKT pathway. Food Funct. 2023, 14, 874–885. [Google Scholar] [CrossRef]
- Xu, H.; Shen, X.; Li, X.; Yang, X.; Chen, C.; Luo, D. The natural product dehydrocurvularin induces apoptosis of gastric cancer cells by activating PARP-1 and caspase-3. Apoptosis 2023, 28, 525–538. [Google Scholar] [CrossRef]
- Meng, J.; Yuan, Y.; Li, Y.; Yuan, B. Effects of hirsuteine on MDA-MB-453 breast cancer cell proliferation. Oncol. Lett. 2022, 25, 4. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.S.; Feng, P.P.; Zhang, Y.Y.; Wang, F.Z.; Wang, G.L.; Fei, H.R. Scutebarbatine A induces ROS-mediated DNA damage and apoptosis in breast cancer cells by modulating MAPK and EGFR/Akt signaling pathway. Chem. Biol. Interact. 2023, 378, 110487. [Google Scholar] [CrossRef]
- Lim, J.S.; Kyung, S.Y.; Jeon, Y.; Kim, I.S.; Kwak, J.H.; Kim, H.S. Anticancer effects of the HDAC inhibitor, 3β,6β-dihydroxyurs-12-en-27-oic acid, in MCF-7 breast cancer cells via the inhibition of Akt/mTOR pathways. Oncol. Rep. 2023, 49, 43. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.M.; Abd El-Karim, S.S.; Mahmoud, A.H.; Amr, A.E.E.; Al-Omar, M.A. A Comparative Study of the Anticancer Activity and PARP-1 Inhibiting Effect of Benzofuran-Pyrazole Scaffold and Its Nano-Sized Particles in Human Breast Cancer Cells. Molecules 2019, 24, 2413. [Google Scholar] [CrossRef] [PubMed]
- Vishwanath, D.; Girimanchanaika, S.S.; Dukanya, D.; Rangappa, S.; Yang, J.R.; Pandey, V.; Lobie, P.E.; Basappa, B. Design and Activity of Novel Oxadiazole Based Compounds That Target Poly(ADP-ribose) Polymerase. Molecules 2022, 27, 703. [Google Scholar] [CrossRef] [PubMed]
- Syam, Y.M.; Anwar, M.M.; Abd El-Karim, S.S.; Elokely, K.M.; Abdelwahed, S.H. New Quinoxaline-Based Derivatives as PARP-1 Inhibitors: Design, Synthesis, Antiproliferative, and Computational Studies. Molecules 2022, 27, 4924. [Google Scholar] [CrossRef]
- Sadeghian, Z.; Bayat, M.; Safari, F. Synthesis and in vitro evaluation of antitumor activity of spiro[indolo[2,1-b]quinazoline-pyrano[2,3-d]pyrimidine] and spiro[indolo[2,1-b]quinazoline-pyrido[2,3-d]pyrimidine] derivatives by using 2D and 3D cell culture models. Mol. Divers. 2022, 26, 3173–3184. [Google Scholar] [CrossRef]
- Evans, D.A. History of the Harvard ChemDraw Project. Angew. Chem. Int. Ed. 2014, 53, 11140–11145. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; De Groot, B.L.; Grubmüller, H. More bang for your buck: Improved use of GPU nodes for GROMACS 2018. J. Comput. Chem. 2019, 40, 2418–2431. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Strain fluctuations and elastic constants. J. Chem. Phys. 1982, 76, 2662–2666. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C. Gnuplot 5.4: An Interactive Plotting Program. 2010. Available online: http://www.gnuplot.info/ (accessed on 20 August 2024).
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inf. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
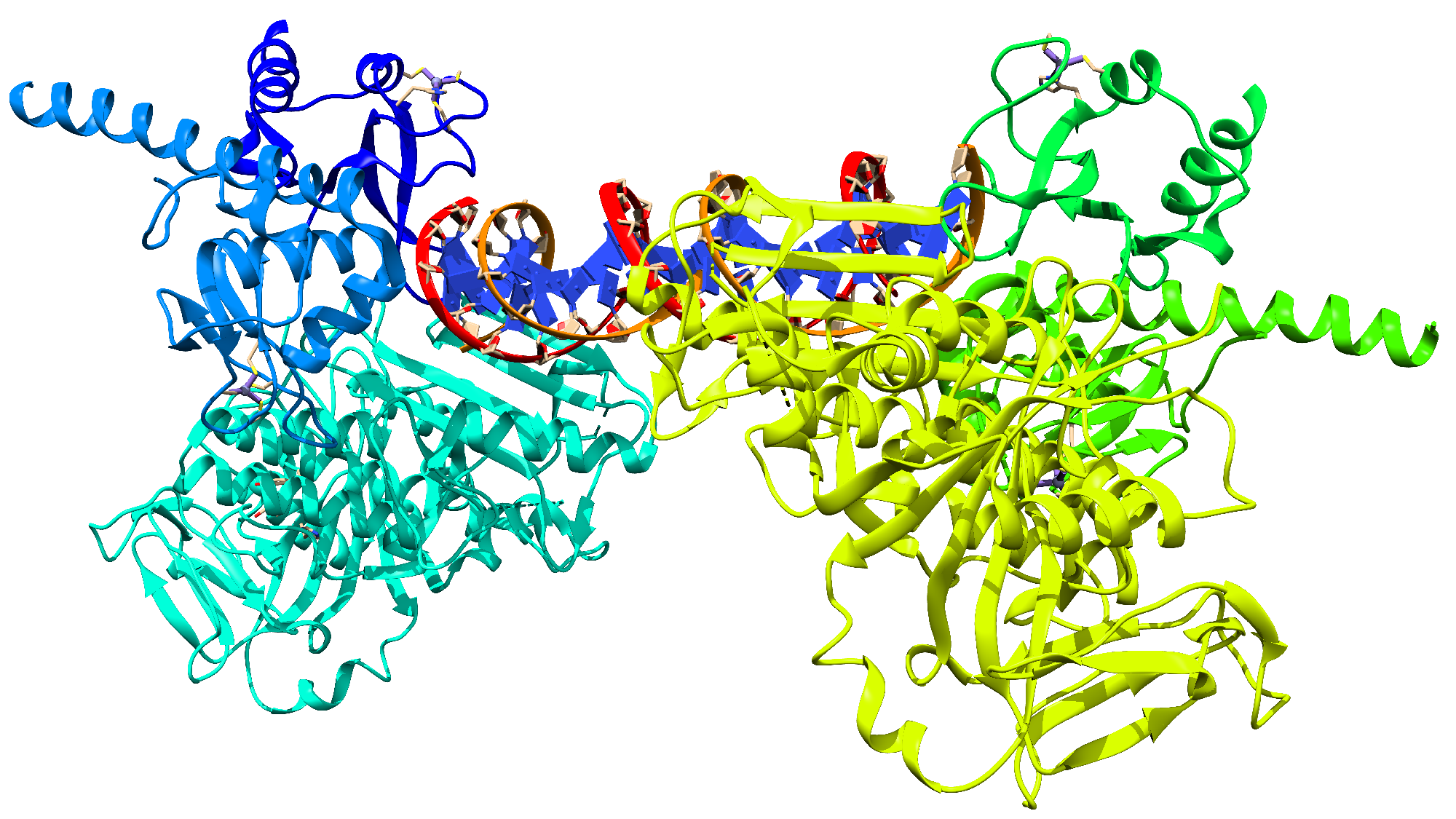
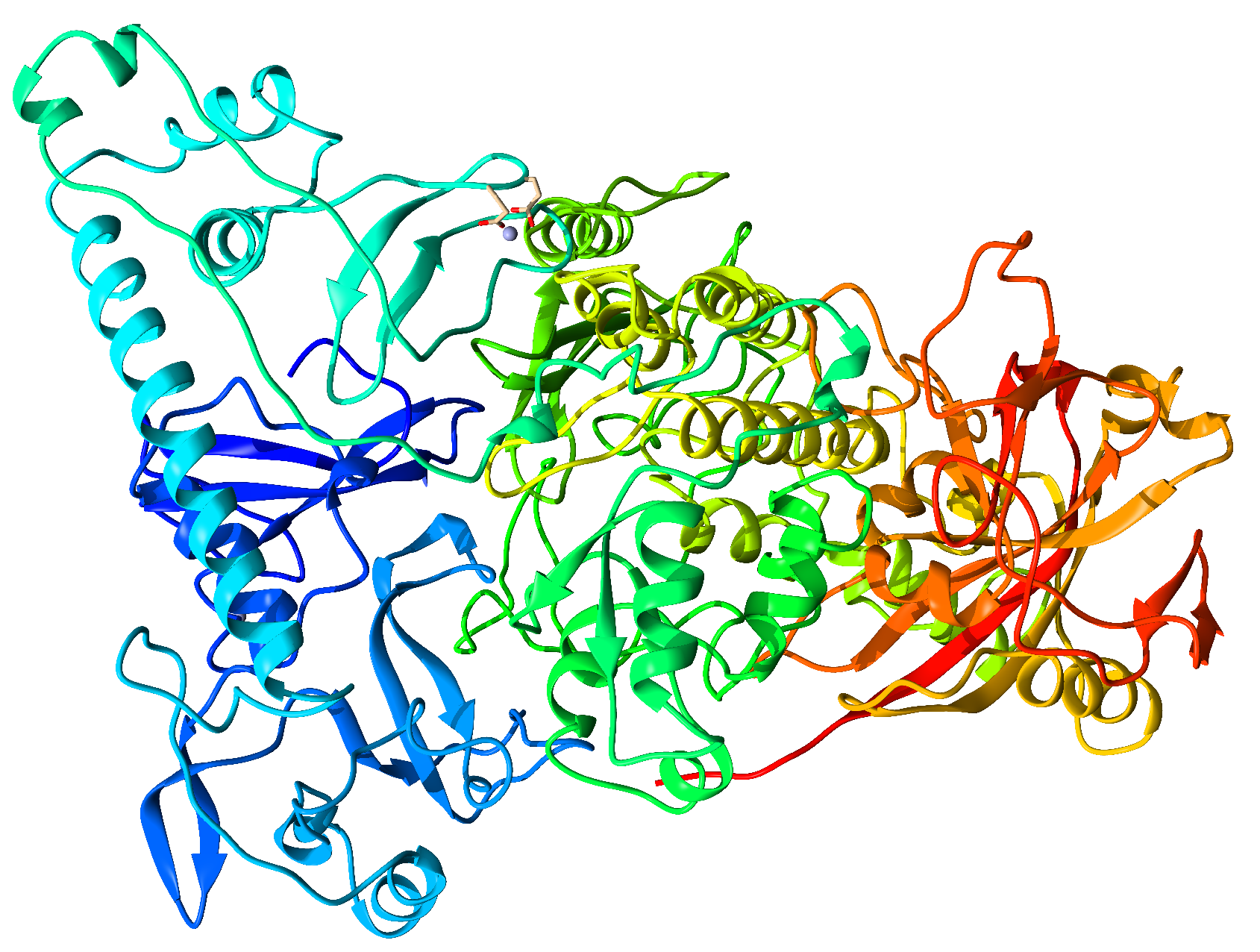
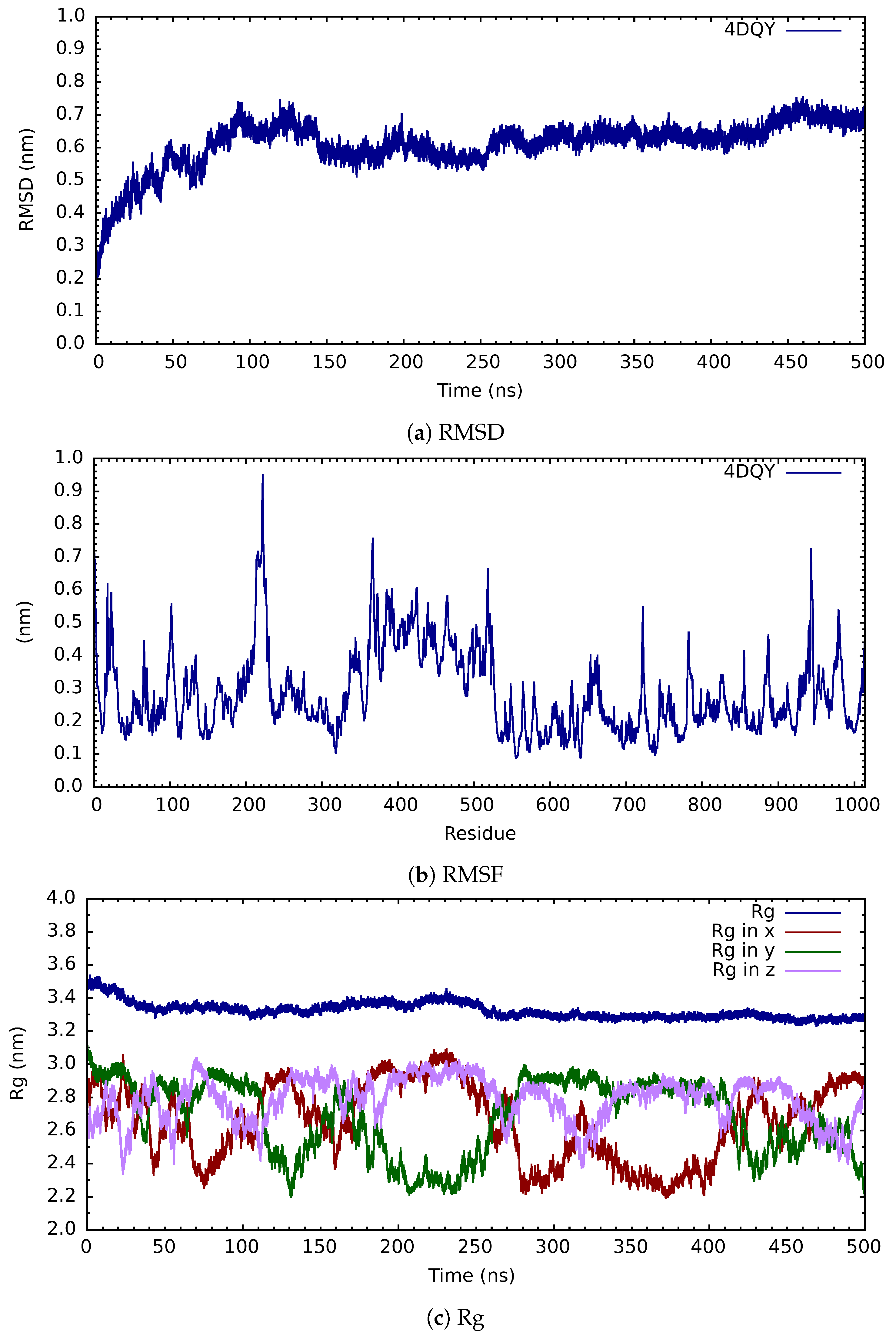
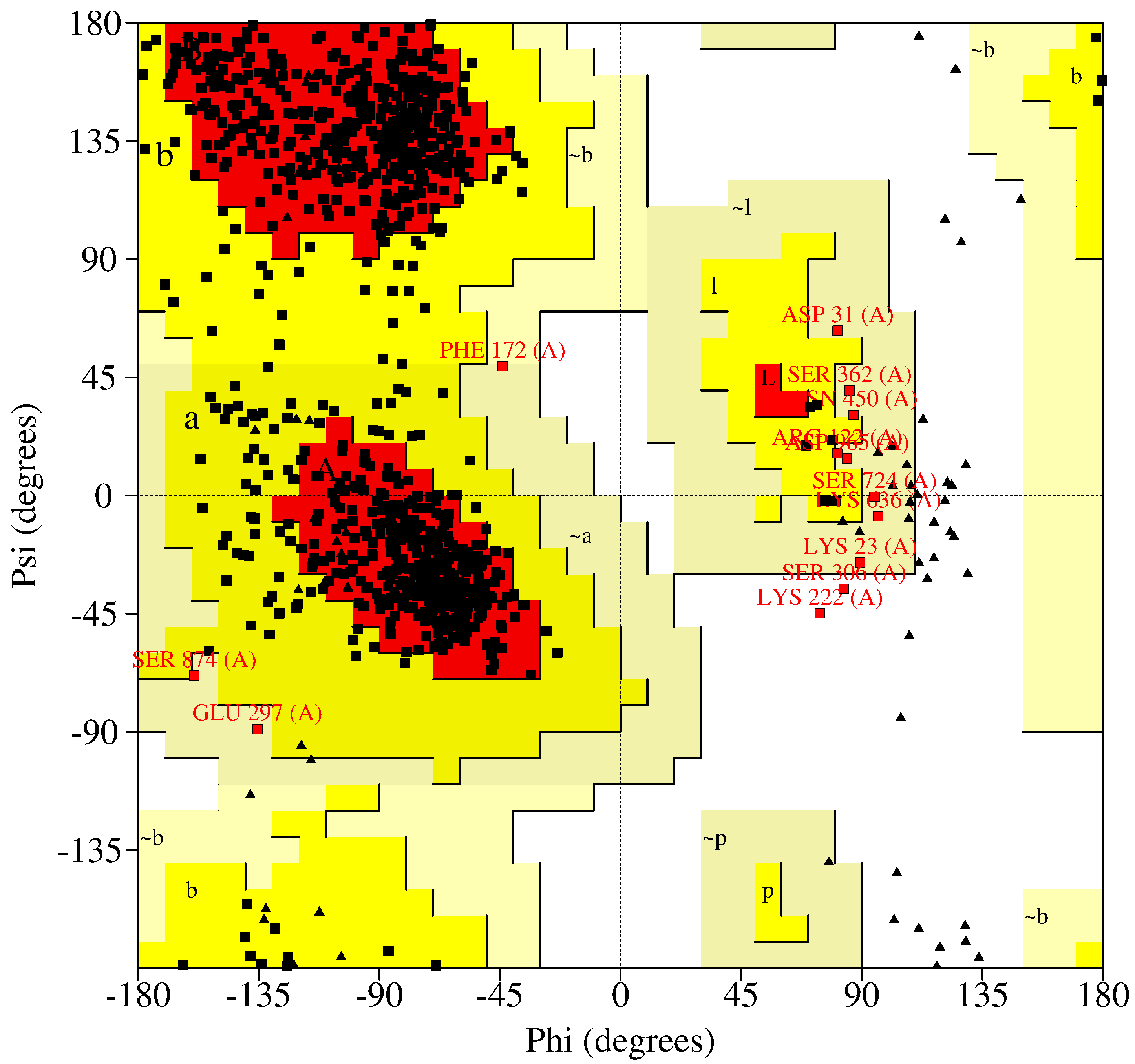
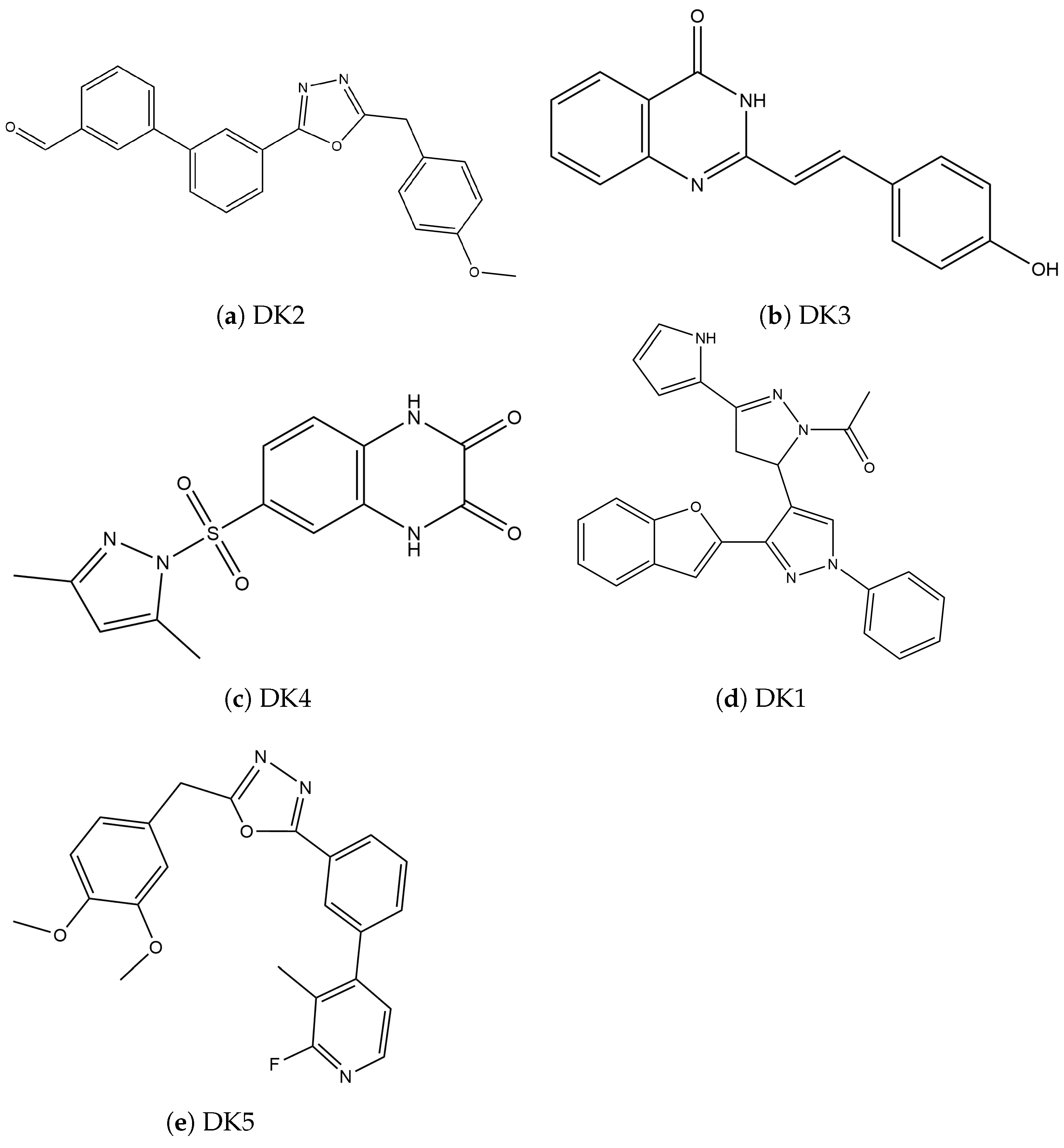
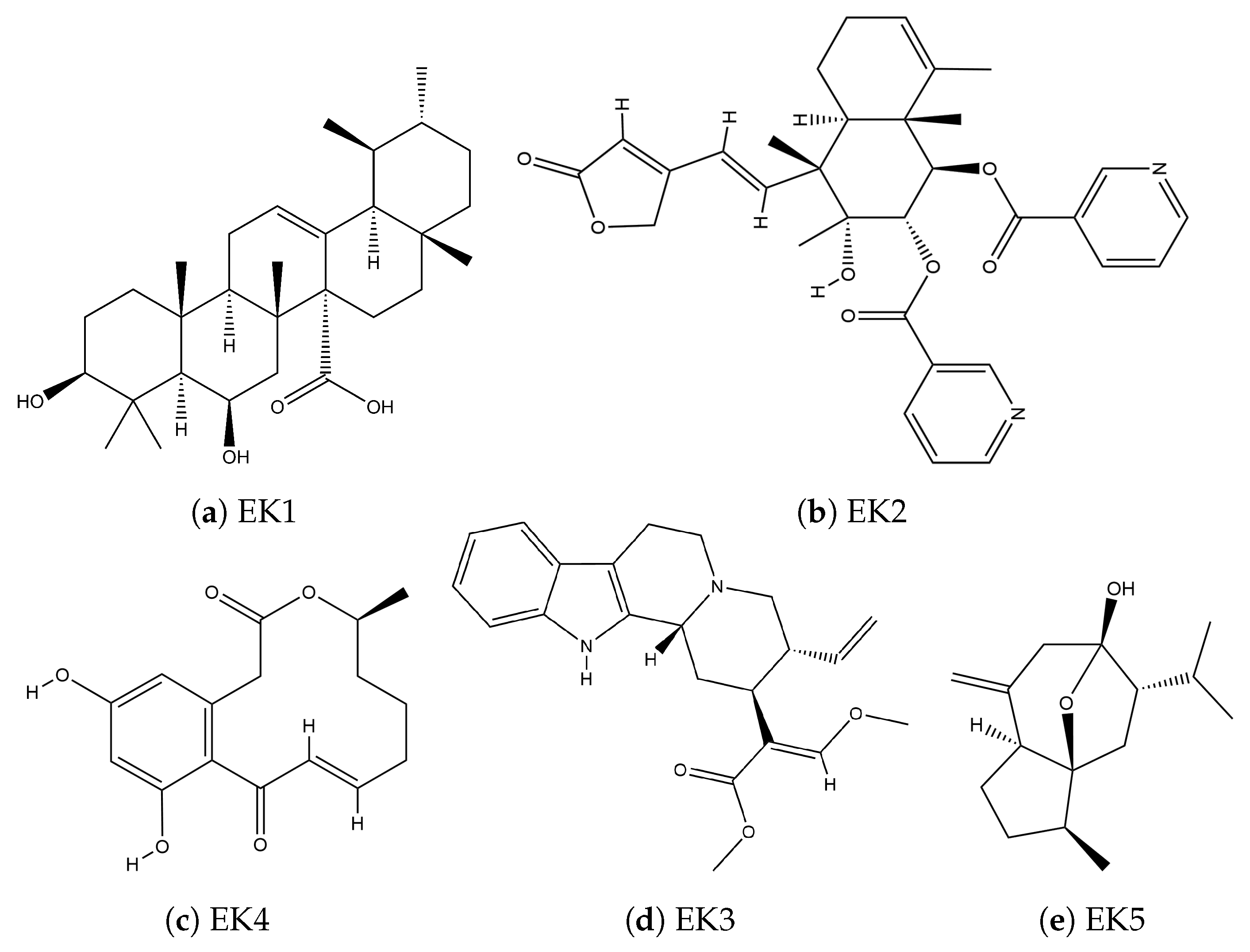
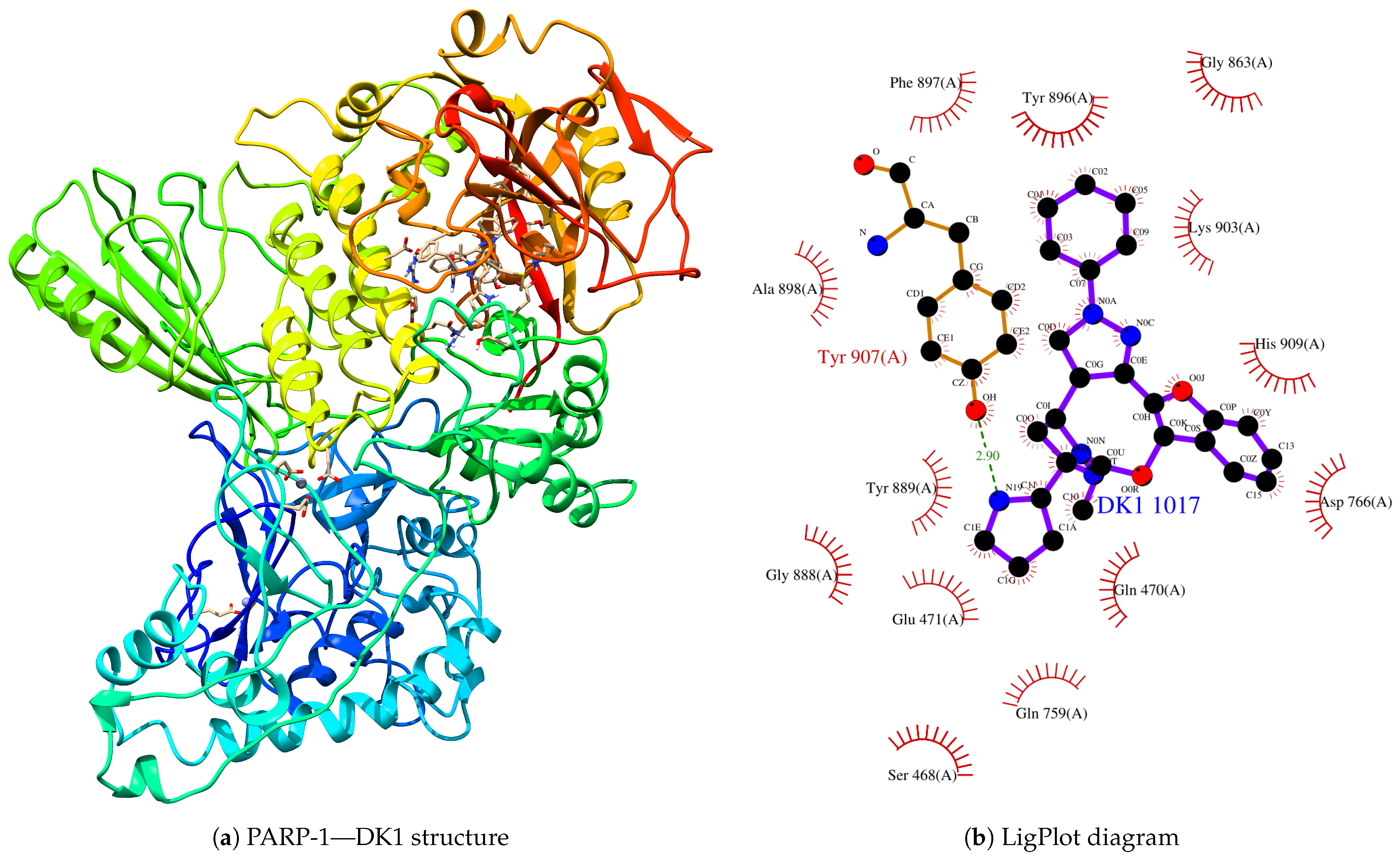
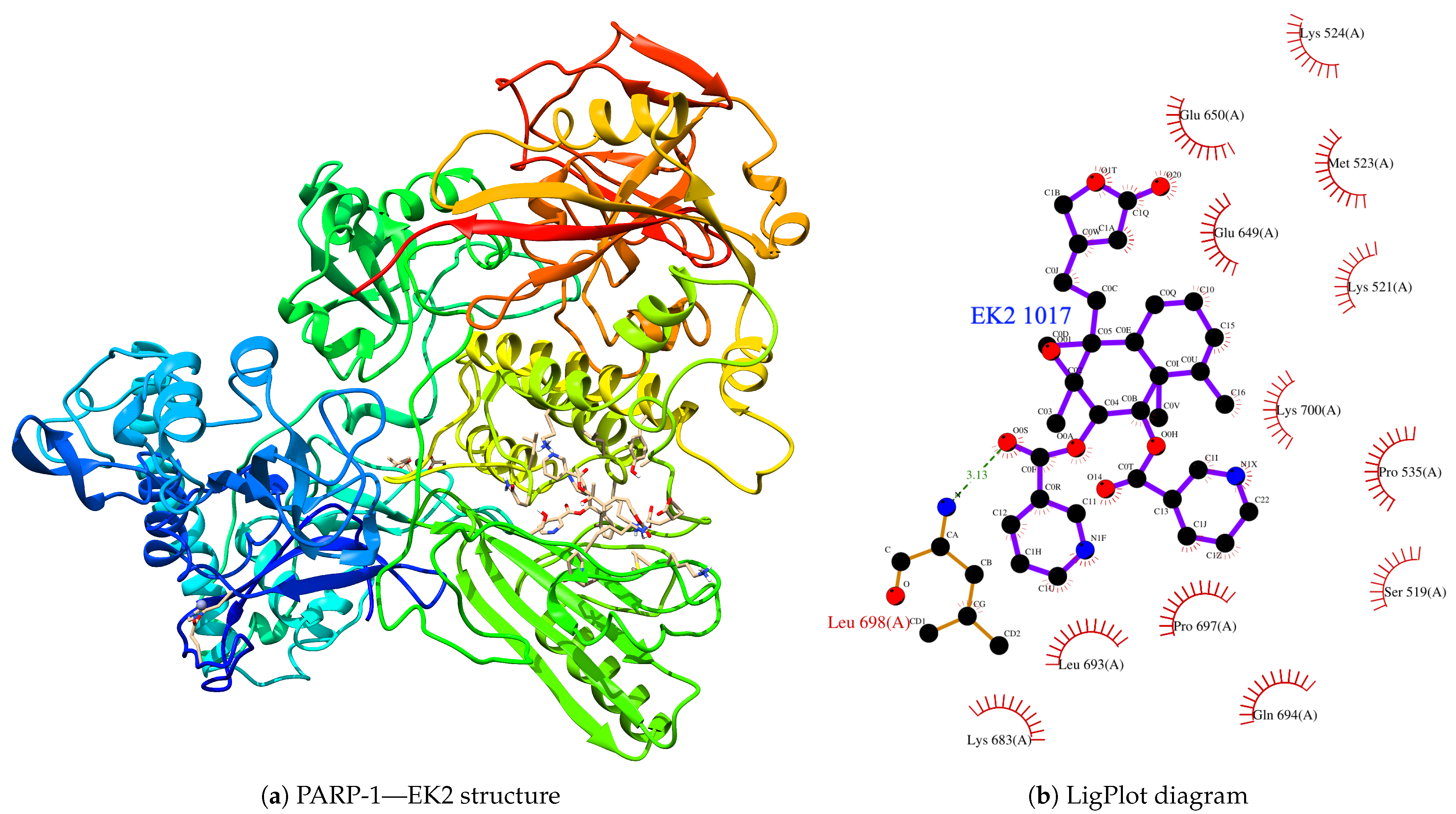


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Inhibitor | Energy |
|---|---|---|
| PARP-1 | DK1 | −9.999 |
| DK2 | −9.573 | |
| DK3 | −9.294 | |
| DK4 | −7.916 | |
| DK5 | −10.260 | |
| PARP-1 | EK1 | −9.997 |
| EK2 | −9.924 | |
| EK3 | −9.182 | |
| EK5 | −9.520 | |
| EK5 | −8.209 |
| Molecule | Binding Energy 1 |
|---|---|
| DK1 | −99.927 ± 6.615 |
| DK2 | −65.858 ± 9.097 |
| DK3 | −63.680 ± 20.180 |
| DK4 | −82.934 ± 6.662 |
| DK5 | −67.150 ± 6.825 |
| EK1 | −65.414 ± 8.264 |
| EK2 | −88.301 ± 6.897 |
| EK3 | −77.123 ± 6.562 |
| EK4 | −40.123 ± 8.044 |
| EK5 | −55.813 ± 5.506 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turpo-Peqqueña, A.G.; Leiva-Flores, E.K.; Luna-Prado, S.; Gómez, B. A Theoretical Study of the Interaction of PARP-1 with Natural and Synthetic Inhibitors: Advances in the Therapy of Triple-Negative Breast Cancer. Curr. Issues Mol. Biol. 2024, 46, 9415-9429. https://doi.org/10.3390/cimb46090558
Turpo-Peqqueña AG, Leiva-Flores EK, Luna-Prado S, Gómez B. A Theoretical Study of the Interaction of PARP-1 with Natural and Synthetic Inhibitors: Advances in the Therapy of Triple-Negative Breast Cancer. Current Issues in Molecular Biology. 2024; 46(9):9415-9429. https://doi.org/10.3390/cimb46090558
Chicago/Turabian StyleTurpo-Peqqueña, Albert Gabriel, Emily Katherine Leiva-Flores, Sebastián Luna-Prado, and Badhin Gómez. 2024. "A Theoretical Study of the Interaction of PARP-1 with Natural and Synthetic Inhibitors: Advances in the Therapy of Triple-Negative Breast Cancer" Current Issues in Molecular Biology 46, no. 9: 9415-9429. https://doi.org/10.3390/cimb46090558