Bioinformatics Approaches to Biomedicine
Share This Topical Collection
Editor
 Dr. Giulia Fiscon
Dr. Giulia Fiscon
Dr. Giulia Fiscon
E-Mail
Website
Collection Editor
Department of Computer, Control, and Management Engineering (DIAG), Sapienza University of Rome, 00179 Rome, Italy
Interests: biomedical engineering; computational biology; bioinformatics; systems biology; computational medicine; network medicine
Special Issues, Collections and Topics in MDPI journals
Topical Collection Information
Dear Colleagues,
Bioinformatics has become an indispensable tool in understanding the biological complexity of diseases and providing unprecedented insights into the biomedical sciences. The growth of omics data allowed studies at different molecular levels of the same sample, aiding to the identification of novel potential biomarkers and therapeutic targets, as well as disease patients’ classification. This Topical Collection aims at presenting computational methods in bioinformatics, along with their application to biomedical sciences.
The contributions may describe novel or and existing approaches to aid: the identification of diagnostic, prognostic, or predictive biomarkers; the discovery of potential therapeutic targets and important disease-related pathways; or provide valuable insights into a precision medicine.
Potential topics include, but are not limited to:
- Biological data mining;
- Omics data analysis (genomics, transcriptomics, proteomics, and metabolomics);
- Omics data integration;
- Modelling Analysis in Health Informatics and Biomedicine;
- Network-based methods and machine learning methods for precision medicine;
- Biomarkers discovery and disease classification.
Dr. Giulia Fiscon
Collection Editor
Manuscript Submission Information
Manuscripts should be submitted online at www.mdpi.com by registering and logging in to this website. Once you are registered, click here to go to the submission form. Manuscripts can be submitted until the deadline. All submissions that pass pre-check are peer-reviewed. Accepted papers will be published continuously in the journal (as soon as accepted) and will be listed together on the collection website. Research articles, review articles as well as short communications are invited. For planned papers, a title and short abstract (about 100 words) can be sent to the Editorial Office for announcement on this website.
Submitted manuscripts should not have been published previously, nor be under consideration for publication elsewhere (except conference proceedings papers). All manuscripts are thoroughly refereed through a single-blind peer-review process. A guide for authors and other relevant information for submission of manuscripts is available on the Instructions for Authors page. Current Issues in Molecular Biology is an international peer-reviewed open access monthly journal published by MDPI.
Please visit the Instructions for Authors page before submitting a manuscript.
The Article Processing Charge (APC) for publication in this open access journal is 2200 CHF (Swiss Francs).
Submitted papers should be well formatted and use good English. Authors may use MDPI's
English editing service prior to publication or during author revisions.
Keywords
- computational medicine
- network theory
- biological data mining
- omics data analysis
- potential therapeutic targets
Published Papers (14 papers)
Open AccessArticle
Bioinformatics Approach to Investigating the Immuno-Inflammatory Mechanisms of Periodontitis in the Progression of Atherosclerosis
by
Wenling Yang, Jianhua Xie, Xing Zhao, Xuelian Li, Qingyi Liu, Jinpeng Sun, Ruiyu Zhang, Yumiao Wei and Boyuan Wang
Viewed by 304
Abstract
Unstable atherosclerotic plaques are a major cause of acute cardiovascular events and ischemic stroke. Clinical studies have suggested a link between periodontitis and atherosclerotic plaque progression, but the underlying mechanisms remain unclear. To investigate this, transcriptomic datasets related to periodontitis and atherosclerosis were
[...] Read more.
Unstable atherosclerotic plaques are a major cause of acute cardiovascular events and ischemic stroke. Clinical studies have suggested a link between periodontitis and atherosclerotic plaque progression, but the underlying mechanisms remain unclear. To investigate this, transcriptomic datasets related to periodontitis and atherosclerosis were downloaded from Gene Expression Omnibus. A weighted gene co-expression network analysis was used to identify gene modules associated with periodontitis, and the Limma R package identified differentially expressed genes (DEGs) between unstable and stable plaques. Overlapping genes were defined as periodontitis-related DEGs, followed by functional enrichment analysis and protein–protein interaction network construction. Machine learning methods were used to identify biomarkers for unstable plaques related to periodontitis, which were validated using external datasets. Immune infiltration and single-cell analyses were performed to explore the relationship between biomarkers and immune cells. A total of 161 periodontitis-related DEGs were identified, with the pathway analysis showing associations with immune regulation and collagen matrix degradation.
HCK,
NCKAP1L, and
WAS were identified as biomarkers for unstable plaques, demonstrating a high diagnostic value (AUC: 0.9884, 95% CI: 0.9641–1). Immune infiltration analysis revealed an increase in macrophages within unstable plaques. Single-cell analysis showed
HCK expression in macrophages and dendritic cells, while
NCKAP1L and
WAS were expressed in macrophages, dendritic cells, NK cells, and T cells. Consensus clustering identified three expression patterns within unstable plaques. Our findings were validated in atherosclerotic mouse models with periodontitis. This study provides insights into how periodontitis contributes to plaque instability, supporting diagnosis and intervention in patients with periodontitis.
Full article
►▼
Show Figures
Open AccessArticle
Unravelling Convergent Signaling Mechanisms Underlying the Aging-Disease Nexus Using Computational Language Analysis
by
Marina Junyent, Haki Noori, Robin De Schepper, Shanna Frajdenberg, Razan Khalid Abdullah Hussen Elsaigh, Patricia H. McDonald, Derek Duckett and Stuart Maudsley
Viewed by 454
Abstract
Multiple lines of evidence suggest that multiple pathological conditions and diseases that account for the majority of human mortality are driven by the molecular aging process. At the cellular level, aging can largely be conceptualized to comprise the progressive accumulation of molecular damage,
[...] Read more.
Multiple lines of evidence suggest that multiple pathological conditions and diseases that account for the majority of human mortality are driven by the molecular aging process. At the cellular level, aging can largely be conceptualized to comprise the progressive accumulation of molecular damage, leading to resultant cellular dysfunction. As many diseases, e.g., cancer, coronary heart disease, Chronic obstructive pulmonary disease, Type II diabetes mellitus, or chronic kidney disease, potentially share a common molecular etiology, then the identification of such mechanisms may represent an ideal locus to develop targeted prophylactic agents that can mitigate this disease-driving mechanism. Here, using the input of artificial intelligence systems to generate unbiased disease and aging mechanism profiles, we have aimed to identify key signaling mechanisms that may represent new disease-preventing signaling pathways that are ideal for the creation of disease-preventing chemical interventions. Using a combinatorial informatics approach, we have identified a potential critical mechanism involving the recently identified kinase, Dual specificity tyrosine-phosphorylation-regulated kinase 3 (DYRK3) and the epidermal growth factor receptor (EGFR) that may function as a regulator of the pathological transition of health into disease via the control of cellular fate in response to stressful insults.
Full article
►▼
Show Figures
Open AccessArticle
Genetic Predisposition to Prediabetes in the Kazakh Population
by
Gulnara Svyatova, Galina Berezina, Alexandra Murtazaliyeva, Altay Dyussupov, Tatyana Belyayeva, Raida Faizova and Azhar Dyussupova
Viewed by 1133
Abstract
The aim of this study was to conduct a comparative analysis of the population frequencies of the minor allele of polymorphic variants in the genes
TCF7L2 (rs7903146) and
PPARG (rs1801282), based on the genome-wide association studies analysis data associated with the risk of
[...] Read more.
The aim of this study was to conduct a comparative analysis of the population frequencies of the minor allele of polymorphic variants in the genes
TCF7L2 (rs7903146) and
PPARG (rs1801282), based on the genome-wide association studies analysis data associated with the risk of developing prediabetes, in an ethnically homogeneous Kazakh population compared to previously studied populations worldwide. This study utilized a genomic database consisting of 1800 ethnically Kazakh individuals who were considered in healthy condition. Whole-genome genotyping was performed using Illumina OmniChip 2.5–8 arrays, which interrogated approximately 2.5 million single nucleotide polymorphisms. The distribution of genotypes for the
TCF7L2 (rs7903146) and
PPARG (rs1801282) polymorphisms in the Kazakh sample was found to be in Hardy–Weinberg equilibrium (
p > 0.05). The minor G allele of the “Asian” protective polymorphism rs1801282 in the
PPARG gene was observed at a frequency of 13.8% in the Kazakh population. This suggests a potentially more significant protective effect of this polymorphism in reducing the risk of prediabetes among Kazakhs. The frequency of the unfavorable T allele of the insulin secretion-disrupting gene
TCF7L2 (rs7903146) in Kazakhs was 15.2%. Studying the associations of genetic markers for prediabetes enables the timely identification of “high-risk groups” and facilitates the implementation of effective preventive measures. Further results from replicative genomic research will help identify significant polymorphic variants of genes underlying the alteration of prediabetes status.
Full article
Open AccessArticle
A Theoretical Study of the Interaction of PARP-1 with Natural and Synthetic Inhibitors: Advances in the Therapy of Triple-Negative Breast Cancer
by
Albert Gabriel Turpo-Peqqueña, Emily Katherine Leiva-Flores, Sebastián Luna-Prado and Badhin Gómez
Viewed by 1737
Abstract
In the current study, we have investigated the secondary metabolites present in ethnomedical plants used for medicinal purposes—Astilbe chinensis (EK1), Scutellaria barbata D. Don (EK2), Uncaria rhynchophylla (EK3), Fallugia paradoxa (EK4), and Curcuma zedoaria (Christm.) Thread (EK5)—and we have compared them with five
[...] Read more.
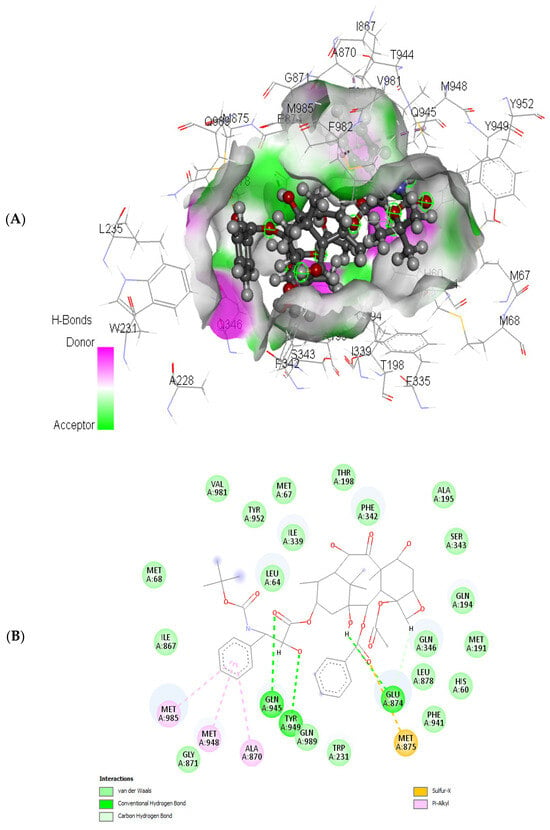
In the current study, we have investigated the secondary metabolites present in ethnomedical plants used for medicinal purposes—Astilbe chinensis (EK1), Scutellaria barbata D. Don (EK2), Uncaria rhynchophylla (EK3), Fallugia paradoxa (EK4), and Curcuma zedoaria (Christm.) Thread (EK5)—and we have compared them with five compounds of synthetic origin for the inhibition of PARP-1, which is linked to abnormal DNA replication, generating carcinogenic cells. We have studied these interactions through molecular dynamics simulations of each interacting system under physiological conditions (pH, temperature, and pressure) and determined that the compounds of natural origin have a capacity to inhibit PARP-1 (Poly(ADP-ribose) Polymerase 1) in all the cases inspected in this investigation. However, it is essential to mention that their interaction energy is relatively lower compared to that of compounds of synthetic origin. Given that binding energy is mandatory for the generation of a scale or classification of which is the best interacting agent, we can say that we assume that compounds of natural origin, having a complexation affinity with PARP-1, induce cell apoptosis, a potential route for the prevention of the proliferation of carcinogenic cells.
Full article
►▼
Show Figures
Open AccessCommunication
Genetic Variants Associated with Body Mass Index Changes in Korean Adults: The Anseong and Ansan Cohorts of the Korean Genome and Epidemiology Study
by
Sang-Im Lee, Su-Kang Kim and Sang-Wook Kang
Viewed by 1187
Abstract
Although previous studies have examined the relationship between obesity and genetics in response to the growing obesity epidemic, research on the relationship between obesity and long-term changes in body mass index (BMI) is limited. To investigate this relationship, data from 1030 cases in
[...] Read more.
Although previous studies have examined the relationship between obesity and genetics in response to the growing obesity epidemic, research on the relationship between obesity and long-term changes in body mass index (BMI) is limited. To investigate this relationship, data from 1030 cases in the Anseong and Ansan cohorts were collected from the Korean Genome and Epidemiology Study conducted by the Korea National Institute of Health between 2000 and 2014. Cases lacking participants’ BMI data throughout the study were excluded, resulting in a final sample size of 3074. An increase or decrease in BMI was analyzed using PLINK, STRING, and DAVID, with significant differences observed in the
AEN,
ANKS1B,
CSF1,
EEF2K,
FRAS1,
GRIK4,
PDGFC,
THTPA, and
TREH genes. These genes were observed to cluster with pathways related to type 2 diabetes, cardiovascular disease, metabolic processes, and endocytosis-related genes. These results suggest that several genes are involved in BMI changes and that several pathways are associated with obesity risk. Moreover, some genetic variants appear to influence BMI changes in Korean adults.
Full article
►▼
Show Figures
Open AccessArticle
A Bioinformatic Analysis Predicts That Cannabidiol Could Function as a Potential Inhibitor of the MAPK Pathway in Colorectal Cancer
by
Julianne du Plessis, Aurelie Deroubaix, Aadilah Omar and Clement Penny
Cited by 3 | Viewed by 1307
Abstract
Colorectal cancer (CRC), found in the intestinal tract, is initiated and progresses through various mechanisms, including the dysregulation of signaling pathways. Several signaling pathways, such as EGFR and MAPK, involved in cell proliferation, migration, and apoptosis, are often dysregulated in CRC. Although cannabidiol
[...] Read more.
Colorectal cancer (CRC), found in the intestinal tract, is initiated and progresses through various mechanisms, including the dysregulation of signaling pathways. Several signaling pathways, such as EGFR and MAPK, involved in cell proliferation, migration, and apoptosis, are often dysregulated in CRC. Although cannabidiol (CBD) has previously induced apoptosis and cell cycle arrest in vitro in CRC cell lines, its effects on signaling pathways have not yet been determined. An in silico analysis was used here to assess partner proteins that can bind to CBD, and docking simulations were used to predict precisely where CBD would bind to these selected proteins. A survey of the current literature was used to hypothesize the effect of CBD binding on such proteins. The results predict that CBD could interact with EGFR, RAS/RAF isoforms, MEK1/2, and ERK1/2. The predicted CBD-induced inhibition might be due to CBD binding to the ATP binding site of the target proteins. This prevents the required phosphoryl transfer to activate substrate proteins and/or CBD binding to the DFG motif from taking place, thus reducing catalytic activity.
Full article
►▼
Show Figures
Open AccessArticle
Using Copy Number Variation Data and Neural Networks to Predict Cancer Metastasis Origin Achieves High Area under the Curve Value with a Trade-Off in Precision
by
Michel-Edwar Mickael, Norwin Kubick, Atanas G. Atanasov, Petr Martinek, Jarosław Olav Horbańczuk, Nikko Floretes, Michael Michal, Tomas Vanecek, Justyna Paszkiewicz, Mariusz Sacharczuk and Piotr Religa
Viewed by 1121
Abstract
The accurate identification of the primary tumor origin in metastatic cancer cases is crucial for guiding treatment decisions and improving patient outcomes. Copy number alterations (CNAs) and copy number variation (CNV) have emerged as valuable genomic markers for predicting the origin of metastases.
[...] Read more.
The accurate identification of the primary tumor origin in metastatic cancer cases is crucial for guiding treatment decisions and improving patient outcomes. Copy number alterations (CNAs) and copy number variation (CNV) have emerged as valuable genomic markers for predicting the origin of metastases. However, current models that predict cancer type based on CNV or CNA suffer from low AUC values. To address this challenge, we employed a cutting-edge neural network approach utilizing a dataset comprising CNA profiles from twenty different cancer types. We developed two workflows: the first evaluated the performance of two deep neural networks—one ReLU-based and the other a 2D convolutional network. In the second workflow, we stratified cancer types based on anatomical and physiological classifications, constructing shallow neural networks to differentiate between cancer types within the same cluster. Both approaches demonstrated high AUC values, with deep neural networks achieving a precision of 60%, suggesting a mathematical relationship between CNV type, location, and cancer type. Our findings highlight the potential of using CNA/CNV to aid pathologists in accurately identifying cancer origins with accessible clinical tests.
Full article
►▼
Show Figures
Open AccessArticle
Application of Transcriptome-Based Gene Set Featurization for Machine Learning Model to Predict the Origin of Metastatic Cancer
by
Yeonuk Jeong, Jinah Chu, Juwon Kang, Seungjun Baek, Jae-Hak Lee, Dong-Sub Jung, Won-Woo Kim, Yi-Rang Kim, Jihoon Kang and In-Gu Do
Cited by 2 | Viewed by 1363
Abstract
Identifying the primary site of origin of metastatic cancer is vital for guiding treatment decisions, especially for patients with cancer of unknown primary (CUP). Despite advanced diagnostic techniques, CUP remains difficult to pinpoint and is responsible for a considerable number of cancer-related fatalities.
[...] Read more.
Identifying the primary site of origin of metastatic cancer is vital for guiding treatment decisions, especially for patients with cancer of unknown primary (CUP). Despite advanced diagnostic techniques, CUP remains difficult to pinpoint and is responsible for a considerable number of cancer-related fatalities. Understanding its origin is crucial for effective management and potentially improving patient outcomes. This study introduces a machine learning framework, ONCOfind-AI, that leverages transcriptome-based gene set features to enhance the accuracy of predicting the origin of metastatic cancers. We demonstrate its potential to facilitate the integration of RNA sequencing and microarray data by using gene set scores for characterization of transcriptome profiles generated from different platforms. Integrating data from different platforms resulted in improved accuracy of machine learning models for predicting cancer origins. We validated our method using external data from clinical samples collected through the Kangbuk Samsung Medical Center and Gene Expression Omnibus. The external validation results demonstrate a top-1 accuracy ranging from 0.80 to 0.86, with a top-2 accuracy of 0.90. This study highlights that incorporating biological knowledge through curated gene sets can help to merge gene expression data from different platforms, thereby enhancing the compatibility needed to develop more effective machine learning prediction models.
Full article
►▼
Show Figures
Open AccessArticle
Multiomics Analysis of the PHLDA Gene Family in Different Cancers and Their Clinical Prognostic Value
by
Safia Iqbal, Md. Rezaul Karim, Shahnawaz Mohammad, Ramya Mathiyalagan, Md. Niaj Morshed, Deok-Chun Yang, Hyocheol Bae, Esrat Jahan Rupa and Dong Uk Yang
Cited by 1 | Viewed by 1400
Abstract
The PHLDA (pleckstrin homology-like domain family) gene family is popularly known as a potential biomarker for cancer identification, and members of the PHLDA family have become considered potentially viable targets for cancer treatments. The PHLDA gene family consists of PHLDA1, PHLDA2, and PHLDA3.
[...] Read more.
The PHLDA (pleckstrin homology-like domain family) gene family is popularly known as a potential biomarker for cancer identification, and members of the PHLDA family have become considered potentially viable targets for cancer treatments. The PHLDA gene family consists of PHLDA1, PHLDA2, and PHLDA3. The predictive significance of PHLDA genes in cancer remains unclear. To determine the role of pleckstrin as a prognostic biomarker in human cancers, we conducted a systematic multiomics investigation. Through various survival analyses, pleckstrin expression was evaluated, and their predictive significance in human tumors was discovered using a variety of online platforms. By analyzing the protein–protein interactions, we also chose a collection of well-known functional protein partners for pleckstrin. Investigations were also carried out on the relationship between pleckstrins and other cancers regarding mutations and copy number alterations. The cumulative impact of pleckstrin and their associated genes on various cancers, Gene Ontology (GO), and pathway analyses were used for their evaluation. Thus, the expression profiles of PHLDA family members and their prognosis in various cancers may be revealed by this study. During this multiomics analysis, we found that among the PHLDA family, PHLDA1 may be a therapeutic target for several cancers, including kidney, colon, and brain cancer, while PHLDA2 can be a therapeutic target for cancers of the colon, esophagus, and pancreas. Additionally, PHLDA3 may be a useful therapeutic target for ovarian, renal, and gastric cancer.
Full article
►▼
Show Figures
Open AccessArticle
Exome Sequencing for the Diagnostics of Osteogenesis Imperfecta in Six Russian Patients
by
Yulia S. Koshevaya, Mariia E. Turkunova, Anastasia O. Vechkasova, Elena A. Serebryakova, Maxim Yu. Donnikov, Svyatoslav I. Papanov, Alexander N. Chernov, Lev N. Kolbasin, Lyudmila V. Kovalenko, Andrey S. Glotov and Oleg S. Glotov
Viewed by 1393
Abstract
Osteogenesis imperfecta (OI) is a group of inherited disorders of connective tissue that cause significant deformities and fragility in bones. Most cases of OI are associated with pathogenic variants in collagen type I genes and are characterized by pronounced polymorphisms in clinical manifestations
[...] Read more.
Osteogenesis imperfecta (OI) is a group of inherited disorders of connective tissue that cause significant deformities and fragility in bones. Most cases of OI are associated with pathogenic variants in collagen type I genes and are characterized by pronounced polymorphisms in clinical manifestations and the absence of clear phenotype–genotype correlation. The objective of this study was to conduct a comprehensive molecular–genetic and clinical analysis to verify the diagnosis of OI in six Russian patients with genetic variants in the
COL1A1 and
COL1A2 genes. Clinical and laboratory data were obtained from six OI patients who were observed at the Medical Genetics Center in Saint Petersburg from 2016 to 2023. Next-generation sequencing on MGISEQ G400 (MGI, China) was used for DNA analysis. The GATK bioinformatic software (version 4.5.0.0) was used for variant calling and hard filtering. Genetic variants were verified by the direct automatic sequencing of PCR products using the ABI 3500X sequencer. We identified six genetic variants, as follows pathogenic c.3505G>A (p. Gly1169Ser), c.769G>A (p.Gly257Arg), VUS c.4123G>A (p.Ala1375Thr), and c.4114A>T (p.Asn1372Tyr) in
COL1A1; and likely pathogenic c.2035G>A (p.Gly679Ser) and c.739-2A>T in
COL1A2. In addition, clinical cases are presented due to the presence of the c.4114A>T variant in the
COL1A2 gene. Molecular genetics is essential for determining different OI types due to the high similarity across various types of the disease and the failure of unambiguous diagnosis based on clinical manifestations alone. Considering the variable approaches to OI classification, an integrated strategy is required for optimal patient management.
Full article
Open AccessArticle
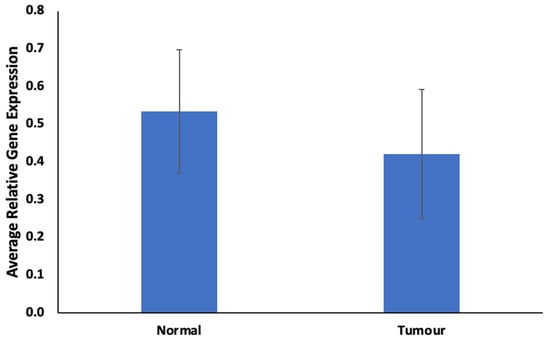
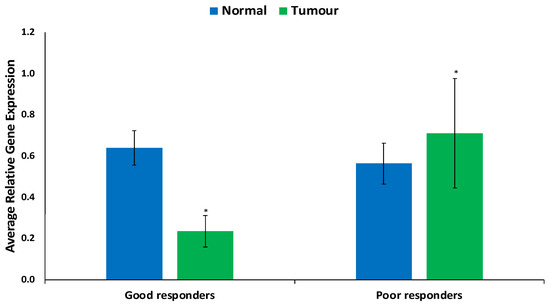
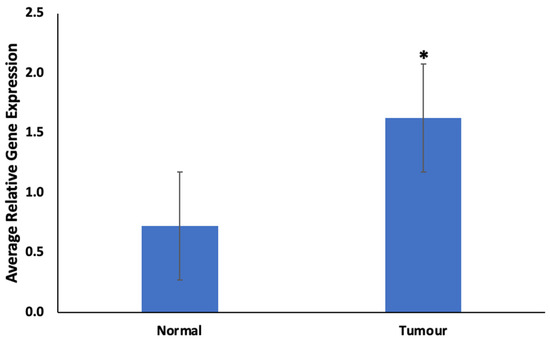
Genetic Signatures for Distinguishing Chemo-Sensitive from Chemo-Resistant Responders in Prostate Cancer Patients
by
Lemohang Gumenku, Mamello Sekhoacha, Beynon Abrahams, Samson Mashele, Aubrey Shoko and Ochuko L. Erukainure
Viewed by 2490
Abstract
Prostate cancer remains a significant public health concern in sub-Saharan Africa, particularly impacting South Africa with high mortality rates. Despite many years of extensive research and significant financial expenditure, there has yet to be a definitive solution to prostate cancer. It is not
[...] Read more.
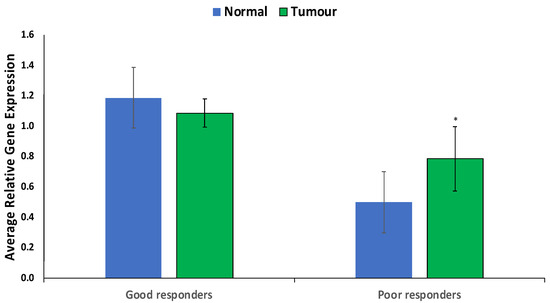
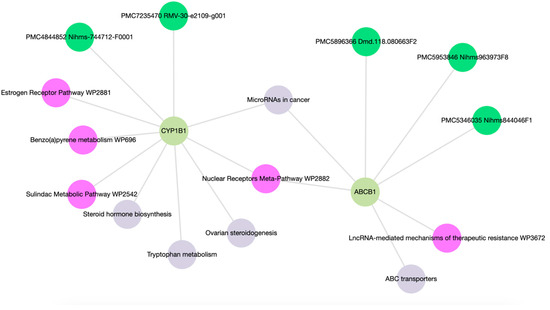
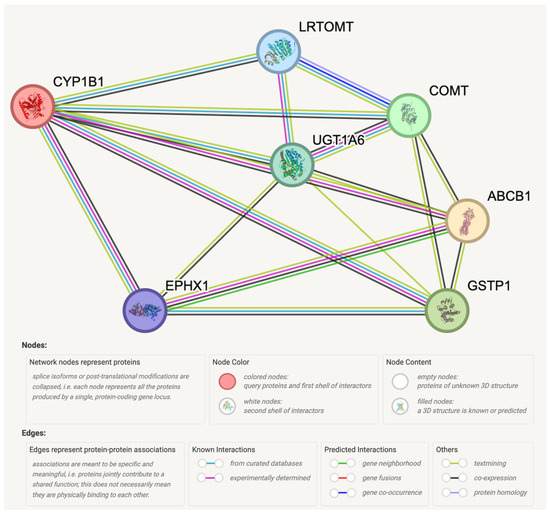
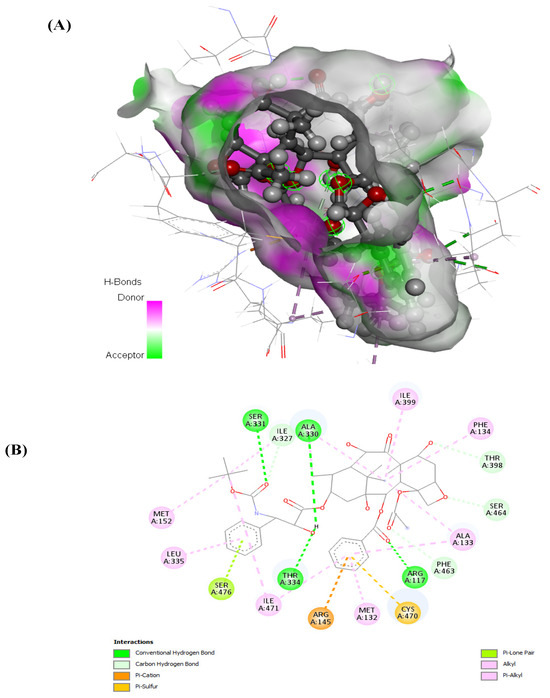
Prostate cancer remains a significant public health concern in sub-Saharan Africa, particularly impacting South Africa with high mortality rates. Despite many years of extensive research and significant financial expenditure, there has yet to be a definitive solution to prostate cancer. It is not just individuals who vary in their response to treatment, but even different nodules within the same tumor exhibit unique transcriptome patterns. These distinctions extend beyond mere differences in gene expression levels to encompass the control and networking of individual genes. Escalating chemotherapy resistance in prostate cancer patients has prompted increased research into its underlying mechanisms. The heterogeneous nature of transcriptomic organization among men makes the pursuit of universal biomarkers and one-size-fits-all treatments impractical. This study delves into the expression of drug resistance-associated genes, ABCB1 and CYP1B1, in cancer cells. Employing bioinformatics, we explored the molecular pathways and cascades linked to drug resistance following upregulation of these genes. Samples were obtained from archived prostate cancer patient specimens through pre-treatment biopsies of two categories: good vs. poor responders, with cDNAs synthesized from isolated RNAs subjected to qPCR analysis. The results revealed increased ABCB1 and CYP1B1 expression in tumor samples of the poor responders. Gene enrichment and network analysis associated ABCB1 with ABC transporters and LncRNA-mediated therapeutic resistance (WP3672), while CYP1B1 was linked to ovarian steroidogenesis, tryptophan metabolism, steroid hormone biosynthesis, benzo(a)pyrene metabolism, the sulindac metabolic pathway, and the estrogen receptor pathway, which are associated with drug resistance. Both ABCB1 and CYP1B1 correlated with microRNAs in cancer and the Nuclear Receptors Meta-Pathway. STRING analysis predicted protein–protein interactions of ABCB1 and CYP1B1 with Glutathione S-transferase Pi, Catechol O-methyltransferase, UDP-glucuronosyltransferase 1-6, Leucine-rich Transmembrane and O-methyltransferase (LRTOMT), and Epoxide hydrolase 1, with scores of 0.973, 0.971, 0.966, 0.966, and 0.966, respectively. Furthermore, molecular docking analysis of the chemotherapy drug, docetaxel, with CYP1B1 and ABCB1 revealed robust molecular interactions, with binding energies of −20.37 and −15.25 Kcal/mol, respectively. These findings underscore the susceptibility of cancer patients to drug resistance due to increased ABCB1 and CYP1B1 expression in tumor samples from patients in the poor-responders category that affects associated molecular pathways. The potent molecular interactions of ABCB1 and CYP1B1 with docetaxel further emphasize the potential basis for chemotherapy resistance.
Full article
►▼
Show Figures
Open AccessArticle
In Silico Approach to Molecular Profiling of the Transition from Ovarian Epithelial Cells to Low-Grade Serous Ovarian Tumors for Targeted Therapeutic Insights
by
Asim Leblebici, Ceren Sancar, Bahar Tercan, Zerrin Isik, Mehmet Emin Arayici, Ender Berat Ellidokuz, Yasemin Basbinar and Nuri Yildirim
Cited by 2 | Viewed by 2347
Abstract
This paper aims to elucidate the differentially coexpressed genes, their potential mechanisms, and possible drug targets in low-grade invasive serous ovarian carcinoma (LGSC) in terms of the biologic continuity of normal, borderline, and malignant LGSC. We performed a bioinformatics analysis, integrating datasets generated
[...] Read more.
This paper aims to elucidate the differentially coexpressed genes, their potential mechanisms, and possible drug targets in low-grade invasive serous ovarian carcinoma (LGSC) in terms of the biologic continuity of normal, borderline, and malignant LGSC. We performed a bioinformatics analysis, integrating datasets generated using the GPL570 platform from different studies from the GEO database to identify changes in this transition, gene expression, drug targets, and their relationships with tumor microenvironmental characteristics. In the transition from ovarian epithelial cells to the serous borderline, the FGFR3 gene in the “Estrogen Response Late” pathway, the ITGB2 gene in the “Cell Adhesion Molecule”, the CD74 gene in the “Regulation of Cell Migration”, and the IGF1 gene in the “Xenobiotic Metabolism” pathway were upregulated in the transition from borderline to LGSC. The ERBB4 gene in “Proteoglycan in Cancer”, the AR gene in “Pathways in Cancer” and “Estrogen Response Early” pathways, were upregulated in the transition from ovarian epithelial cells to LGSC. In addition, SPP1 and ITGB2 genes were correlated with macrophage infiltration in the LGSC group. This research provides a valuable framework for the development of personalized therapeutic approaches in the context of LGSC, with the aim of improving patient outcomes and quality of life. Furthermore, the main goal of the current study is a preliminary study designed to generate in silico inferences, and it is also important to note that subsequent in vitro and in vivo studies will be necessary to confirm the results before considering these results as fully reliable.
Full article
►▼
Show Figures
Open AccessBrief Report
Association between Single Nucleotide Polymorphisms in Monoamine Oxidase and the Severity of Addiction to Betel Quid
by
Chung-Chieh Hung, Ying-Chin Ko and Chia-Min Chung
Cited by 3 | Viewed by 1483
Abstract
Betel quid (BQ) is the fourth most popular psychoactive substance in the world, and BQ use disorder (BUD) is prevalent in Asian countries. Although the mechanisms underlying BUD remain unclear, studies have reported influences from monoamine oxidase inhibitor. We enrolled 50 patients with
[...] Read more.
Betel quid (BQ) is the fourth most popular psychoactive substance in the world, and BQ use disorder (BUD) is prevalent in Asian countries. Although the mechanisms underlying BUD remain unclear, studies have reported influences from monoamine oxidase inhibitor. We enrolled 50 patients with BUD and assessed their BQ consumption habits, emotional conditions, and the clinical severity of addiction—assessed using the
Diagnostic and Statistical Manual of Mental Disorders [Fifth Edition] (
DSM-5) criteria, Substance Use Severity Rating Scale, and Yale–Brown Obsessive Compulsive Disorder Rating Scale for BQ. Patients were categorized into the severe group when showing six or more symptoms defined by
DSM-5. A genome-wide association study was conducted for single nucleotide polymorphisms in
BRCA1,
COL9A1,
NOTCH1,
HSPA13,
FAT1, and
MAOA by using patients’ blood samples. More severe BUD symptoms were associated with younger age of using BQ and poor oral hygiene and with severe craving for and more anxiety toward BQ use. The
MAOA rs5953210 polymorphism was significantly associated with severe BUD (odds ratio, 6.43; 95% confidence interval, 5.12–7.74;
p < 0.01) and might contribute to BQ-associated cancer risk. Further studies are required to investigate the addictive properties of BQ and the development of novel diagnostic tools and pharmacotherapeutic alternatives to BUD treatment.
Full article
Open AccessArticle
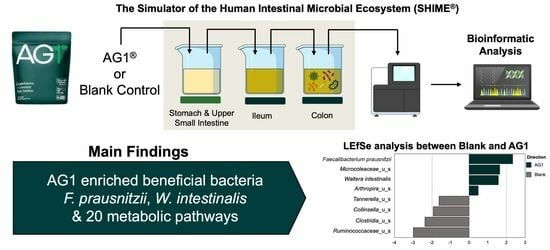
AG1® Induces a Favorable Impact on Gut Microbial Structure and Functionality in the Simulator of Human Intestinal Microbial Ecosystem® Model
by
Trevor O. Kirby, Philip A. Sapp, Jeremy R. Townsend, Marlies Govaert, Cindy Duysburgh, Massimo Marzorati, Tess M. Marshall and Ralph Esposito
Cited by 1 | Viewed by 4189
Abstract
Modulation of the human gut microbiome has become an area of interest in the nutraceutical space. We explored the effect of the novel foundational nutrition supplement AG1
® on the composition of human microbiota in an in vitro experimental design. Employing the Simulator
[...] Read more.
Modulation of the human gut microbiome has become an area of interest in the nutraceutical space. We explored the effect of the novel foundational nutrition supplement AG1
® on the composition of human microbiota in an in vitro experimental design. Employing the Simulator of Human Intestinal Microbial Ecosystem (SHIME
®) model, AG1
® underwent digestion, absorption, and subsequent colonic microenvironment simulation under physiologically relevant conditions in healthy human fecal inocula. Following 48 h of colonic simulation, the gut microbiota were described using shallow shotgun, whole genome sequencing. Metagenomic data were used to describe changes in community structure (alpha diversity, beta diversity, and changes in specific taxa) and community function (functional heterogeneity and changes in specific bacterial metabolic pathways). Results showed no significant change in alpha diversity, but a significant effect of treatment and donor and an interaction between the treatment and donor effect on structural heterogeneity likely stemming from the differential enrichment of eight bacterial taxa. Similar findings were observed for community functional heterogeneity likely stemming from the enrichment of 20 metabolic pathways characterized in the gene ontology term database. It is logical to conclude that an acute dose of AG1 has significant effects on gut microbial composition that may translate into favorable effects in humans.
Full article
►▼
Show Figures












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}