_Kim.png)

Dihydropyrimidine Dehydrogenase Polymorphism c.2194G>A Screening Is a Useful Tool for Decreasing Gastrointestinal and Hematological Adverse Drug Reaction Risk in Fluoropyrimidine-Treated Patients

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Patients
2.2. Statistical Analysis
3. Results
3.1. Characteristics of Patient Candidates for Fluoropyrimidine Therapies
3.2. Analysis of DPYD Polymorphisms in the Cohort

{kind=link}
| Variant | rsID | Nucleotide Change | Amino Acid Change | Dpd Activity | DPYD Carriers (n) (%) | Suggested Dose (5-FU/ Capecitabine etc.) | Allele Frequency Reported in gnomAD | Allele Frequency Observed in the Cohort |
|---|---|---|---|---|---|---|---|---|
| *6 | rs1801160 | c.2194G>A | (p.Val732Ile) | Normal function | 26 (19.70%) | 100% ab initio, 85% in case of toxicity | 0.04675 | 0.065 |
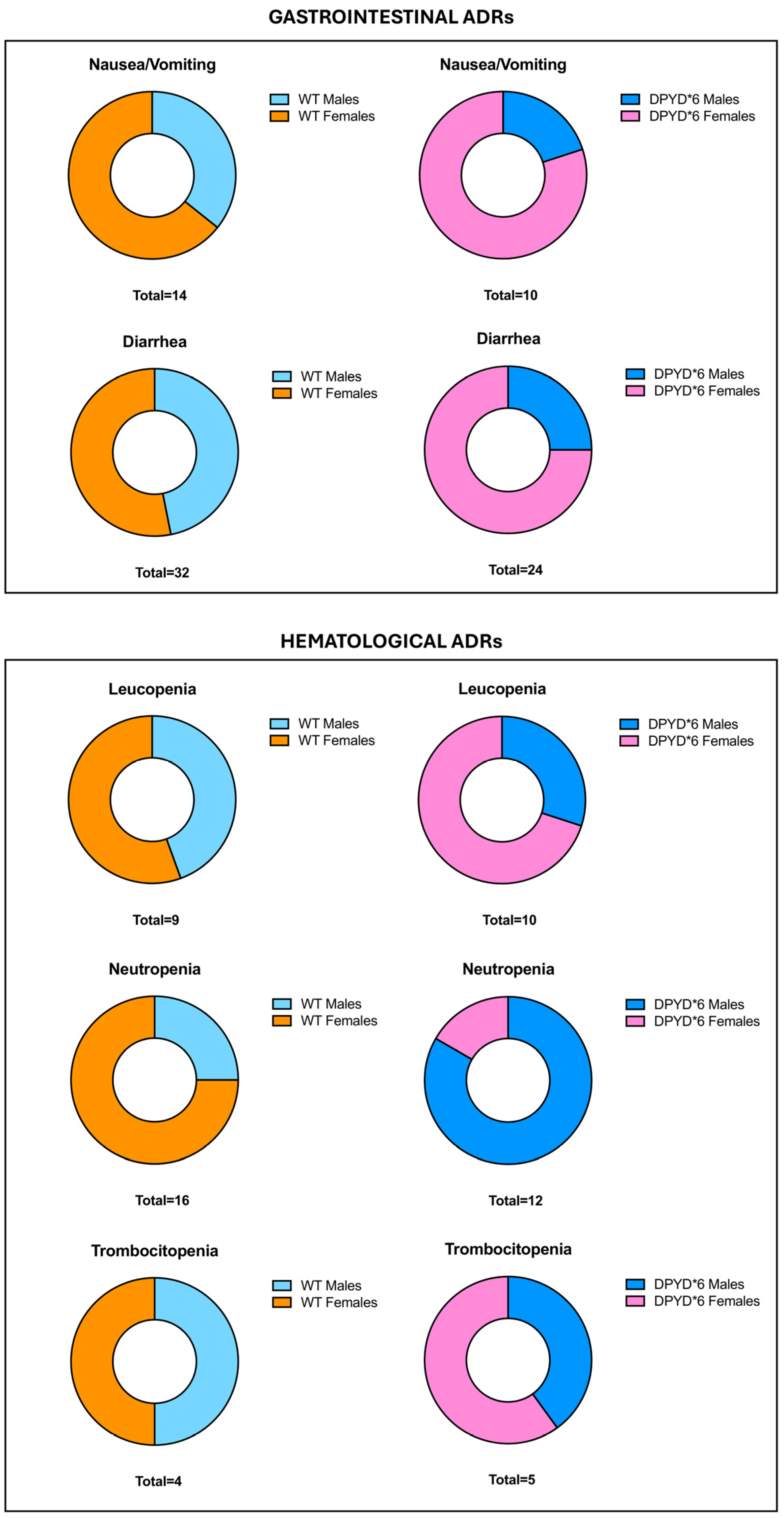
3.3. Reported ADRs after Fluoropyrimidines Treatment in Carrier Patients of WT and DPYD*6
| ADRs Gastrointestinal (76.5%) | Grade ≥ 2 |
|---|---|
| Nausea/Vomiting | 24 (18.18%) |
| Diarrhea | 56 (42.41%) |
| Stomatitis | 21 (15.91%) |
| ADRs Dermatological (8.33%) | Grade ≥ 2 |
| Hand–foot syndrome | 11 (8.33%) |
| ADRs Hematological (52.27%) | Grade ≥ 3 |
| Fever | 6 (4.55%) |
| Leucopenia | 19 (14.39%) |
| Neutropenia | 28 (21.21%) |
| Anemia | 7 (5.30%) |
| Thrombocytopenia | 9 (6.82%) |
| ADRs Neurological (2.94%) | |
| Peripheral neuropathy | 4 (2.94%) |
3.4. Assessment of ADRs between Patients with Mutated DPYD*6 and WT Stratifying by Gender
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marino, P.; Mininni, M.; Deiana, G.; Marino, G.; Divella, R.; Bochicchio, I.; Giuliano, A.; Lapadula, S.; Lettini, A.R.; Sanseverino, F. Healthy Lifestyle and Cancer Risk: Modifiable Risk Factors to Prevent Cancer. Nutrients 2024, 16, 800. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, T.; Ruszkowska, M.; Danielewicz, A.; Niedźwiedzka, E.; Arłukowicz, T.; Przybyłowicz, K.E. A review of colorectal cancer in terms of epidemiology, risk factors, development, symptoms and diagnosis. Cancers 2021, 13, 2025. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef]
- Ardizzone, A.; Basilotta, R.; Filippone, A.; Crupi, L.; Lanza, M.; Lombardo, S.P.; Colarossi, C.; Sciacca, D.; Cuzzocrea, S.; Esposito, E.; et al. Recent Emerging Immunological Treatments for Primary Brain Tumors: Focus on Chemokine-Targeting Immunotherapies. Cells 2023, 12, 841. [Google Scholar] [CrossRef]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.J. Drug resistance in colorectal cancer: General aspects. In Drug Resistance in Colorectal Cancer: Molecular Mechanisms and Therapeutic Strategies; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–33. [Google Scholar]
- Yuan, H.; Zheng, B.a.; Tu, S. Clinical research of intraperitoneal implantation of sustained-release 5-fluorouracil in advanced colorectal cancer. World J. Surg. Oncol. 2015, 13, 320. [Google Scholar] [CrossRef] [PubMed]
- Minchev, V.T. Toxic and Adverse Effects of Chemotherapy with 5-Fluoropyrimidine Drugs. Could Dihydropyrimidine Dehydrogenase Enzyme Screening Serve as a Prerequisite to Successful Chemotherapy? J. Biomed. Clin. Res. 2020, 13, 87–99. [Google Scholar] [CrossRef]
- García-Alfonso, P.; Muñoz Martín, A.J.; Ortega Morán, L.; Soto Alsar, J.; Torres Perez-Solero, G.; Blanco Codesido, M.; Calvo Ferrandiz, P.A.; Grasso Cicala, S. Oral drugs in the treatment of metastatic colorectal cancer. Ther. Adv. Med. Oncol. 2021, 13, 17588359211009001. [Google Scholar] [CrossRef]
- Rapoport, B.L.; Cooksley, T.; Johnson, D.B.; Anderson, R.; Shannon, V.R. Treatment of infections in cancer patients: An update from the neutropenia, infection and myelosuppression study group of the Multinational Association for Supportive Care in Cancer (MASCC). Expert Rev. Clin. Pharmacol. 2021, 14, 295–313. [Google Scholar] [CrossRef]
- Ray, J.; Mahmood, A.; Dogar, M.; Guo, J.; Nwamaghinna, F.; Salciccioli, L.; McFarlane, S.I. Simultaneous cardiotoxicity and neurotoxicity associated with 5-fluorouracil containing chemotherapy: A case report and literature review. Am. J. Med. Case Rep. 2020, 8, 73–75. [Google Scholar] [CrossRef]
- Hishinuma, E.; Gutiérrez Rico, E.; Hiratsuka, M. In Vitro Assessment of Fluoropyrimidine-Metabolizing Enzymes: Dihydropyrimidine Dehydrogenase, Dihydropyrimidinase, and β-Ureidopropionase. J. Clin. Med. 2020, 9, 2342. [Google Scholar] [CrossRef]
- Gmeiner, W.H.; Okechukwu, C.C. Review of 5-FU resistance mechanisms in colorectal cancer: Clinical significance of attenuated on-target effects. Cancer Drug Resist. 2023, 6, 257. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.D.; Pozios, I.; Liu, V.; Nachbichler, S.B.; Böhmer, D.; Kamphues, C.; Beyer, K.; Bruns, C.J.; Kreis, M.E.; Seeliger, H. Thymidine phosphorylase induction by ionizing radiation antagonizes 5-fluorouracil resistance in human ductal pancreatic adenocarcinoma. Radiat. Environ. Biophys. 2022, 61, 255–262. [Google Scholar] [CrossRef]
- Diasio, R.B.; Offer, S.M. Testing for Dihydropyrimidine Dehydrogenase Deficiency to Individualize 5-Fluorouracil Therapy. Cancers 2022, 14, 3207. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.-S.; Huang, C.-Y. Binding Pattern and Structural Interactome of the Anticancer Drug 5-Fluorouracil: A Critical Review. Int. J. Mol. Sci. 2024, 25, 3404. [Google Scholar] [CrossRef]
- Jeong, S.H.; Chavani, O.; Burns, K.; Porter, D.; Findlay, M.; Helsby, N. Severe 5-Fluorouracil-Associated Gastrointestinal Toxicity Unexplained by Dihydropyrimidine Dehydrogenase Deficiency and Renal Impairment: Should We Be Investigating Other Elimination Pathways to Assess the Risk of 5-Fluorouracil Toxicity? Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Gorzkiewicz, M.; Klajnert-Maculewicz, B. Dendrimers as nanocarriers for nucleoside analogues. Eur. J. Pharm. Biopharm. 2017, 114, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Bré, J.; Dickson, A.L.; Read, O.J.; Zhang, Y.; McKissock, F.G.; Mullen, P.; Tang, P.; Zickuhr, G.M.; Czekster, C.M.; Harrison, D.J. The novel anti-cancer fluoropyrimidine NUC-3373 is a potent inhibitor of thymidylate synthase and an effective DNA-damaging agent. Cancer Chemother. Pharmacol. 2023, 91, 401–412. [Google Scholar] [CrossRef]
- De Luca, O.; Salerno, G.; De Bernardini, D.; Torre, M.S.; Simmaco, M.; Lionetto, L.; Gentile, G.; Borro, M. Predicting dihydropyrimidine dehydrogenase deficiency and related 5-fluorouracil toxicity: Opportunities and challenges of DPYD exon sequencing and the role of phenotyping assays. Int. J. Mol. Sci. 2022, 23, 13923. [Google Scholar] [CrossRef]
- Machover, D. A comprehensive review of 5-fluorouracil and leucovorin in patients with metastatic colorectal carcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1997, 80, 1179–1187. [Google Scholar] [CrossRef]
- Capitain, O.; Seegers, V.; Metges, J.-P.; Faroux, R.; Stampfli, C.; Ferec, M.; Budnik, T.M.; Senellart, H.; Rossi, V.; Blouin, N. Comparison of 4 screening methods for detecting fluoropyrimidine toxicity risk: Identification of the most effective, cost-efficient method to save lives. Dose-Response 2020, 18, 1559325820951367. [Google Scholar] [CrossRef]
- Young, D.; Vine, E.; Ghanbarpour, A.; Shani, J.; Siemsen, J.; Wolf, W. Metabolic and distribution studies with radiolabeled 5-fluorouracil. Nukl. Nucl. 1982, 21, 1–7. [Google Scholar] [CrossRef]
- Adam, C.; Bray, T.L.; Pérez-López, A.M.; Tan, E.H.; Rubio-Ruiz, B.; Baillache, D.J.; Houston, D.R.; Salji, M.J.; Leung, H.Y.; Unciti-Broceta, A. A 5-FU precursor designed to evade anabolic and catabolic drug pathways and activated by Pd chemistry in vitro and in vivo. J. Med. Chem. 2022, 65, 552–561. [Google Scholar] [CrossRef]
- Negarandeh, R.; Salehifar, E.; Saghafi, F.; Jalali, H.; Janbabaei, G.; Abdhaghighi, M.J.; Nosrati, A. Evaluation of adverse effects of chemotherapy regimens of 5-fluoropyrimidines derivatives and their association with DPYD polymorphisms in colorectal cancer patients. BMC Cancer 2020, 20, 560. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Elizondo, G.; Sapone, A.; McLeod, H.L.; Raunio, H.; Fernandez-Salguero, P.; Gonzalez, F.J. Characterization of the human dihydropyrimidine dehydrogenase gene. Genomics 1998, 51, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; Cinieri, S.; Michelucci, A.; Salvadori, S.; Loupakis, F.; Schirripa, M.; Cremolini, C.; Crucitta, S.; Barbara, C.; Di Leo, A.; et al. DPYD*6 plays an important role in fluoropyrimidine toxicity in addition to DPYD*2A and c.2846A>T: A comprehensive analysis in 1254 patients. Pharmacogenom. J. 2019, 19, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Bozina, N.; Bilic, I.; Ganoci, L.; Simicevic, L.; Plestina, S.; Lesnjakovic, L.; Trkulja, V. DPYD polymorphisms c.496A>G, c.2194G>A and c.85T>C and risk of severe adverse drug reactions in patients treated with fluoropyrimidine-based protocols. Br. J. Clin. Pharmacol. 2022, 88, 2190–2202. [Google Scholar] [CrossRef]
- De Mattia, E.; Silvestri, M.; Polesel, J.; Ecca, F.; Mezzalira, S.; Scarabel, L.; Zhou, Y.; Roncato, R.; Lauschke, V.M.; Calza, S. Rare genetic variant burden in DPYD predicts severe fluoropyrimidine-related toxicity risk. Biomed. Pharmacother. 2022, 154, 113644. [Google Scholar] [CrossRef]
- Borràs, E.; Dotor, E.; Arcusa, À.; Hernan, I.; de Sousa Dias, M.; Agúndez, J.A.; Carballo, M. High-resolution melting analysis of the common c. 1905+ 1G> A mutation causing dihydropyrimidine dehydrogenase deficiency and lethal 5-fluorouracil toxicity. Front. Genet. 2013, 3, 35356. [Google Scholar] [CrossRef]
- Naushad, S.M.; Hussain, T.; Alrokayan, S.A.; Kutala, V.K. Pharmacogenetic profiling of dihydropyrimidine dehydrogenase (DPYD) variants in the Indian population. J. Gene Med. 2021, 23, e3289. [Google Scholar] [CrossRef]
- Crucitta, S.; Sciandra, F.; Cerbioni, A.; Cucchiara, F.; Arici, R.; Rafaniello, C.; Capuano, A.; van Schaik, R.; Danesi, R.; Del Re, M. Safety matters: The troubled and finally successful story of dihydropyrimidine dehydrogenase pharmacogenetic test in cancer patients. Ann. Res. Oncol. 2021, 1, 71–84. [Google Scholar] [CrossRef]
- Bolufer, I.F.; Vallés, X.G.; Álvarez, S.I.; Mira, A.S.; Luján, C.G.; Aguilera, M.J.S.; Tarancón, R.G.; Naranjo, M.B.; Salas, P.C.; González, M.S. Diversity of oncopharmacogenetic profile within Spanish population. Pharmacogenet. Genom. 2024, 34, 166–169. [Google Scholar] [CrossRef]
- Medwid, S.; Wigle, T.J.; Kim, R.B. Fluoropyrimidine-associated toxicity and DPYD variants c. 85T> C, c. 496A> G, and c. 1236G> A: Impact of haplotype. Cancer Chemother. Pharmacol. 2023, 91, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.F.; Turner, R.M.; Pirmohamed, M. Pharmacogenomics of anticancer drugs: Personalising the choice and dose to manage drug response. Br. J. Clin. Pharmacol. 2021, 87, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Koo, K.; Pasternak, A.L.; Henry, N.L.; Sahai, V.; Hertz, D.L. Survey of US medical oncologists’ practices and beliefs regarding DPYD testing before fluoropyrimidine chemotherapy. JCO Oncol. Pract. 2022, 18, e958–e965. [Google Scholar] [CrossRef]
- Lunenburg, C.A.; van Staveren, M.C.; Gelderblom, H.; Guchelaar, H.-J.; Swen, J.J. Evaluation of clinical implementation of prospective DPYD genotyping in 5-fluorouracil-or capecitabine-treated patients. Pharmacogenomics 2016, 17, 721–729. [Google Scholar] [CrossRef]
- Peruzzi, E.; Roncato, R.; De Mattia, E.; Bignucolo, A.; Swen, J.J.; Guchelaar, H.J.; Toffoli, G.; Cecchin, E. Implementation of pre-emptive testing of a pharmacogenomic panel in clinical practice: Where do we stand? Br. J. Clin. Pharmacol. 2023; early view. [Google Scholar]
- Soria-Chacartegui, P.; Villapalos-Garcia, G.; Lopez-Fernandez, L.A.; Navares-Gomez, M.; Mejia-Abril, G.; Abad-Santos, F.; Zubiaur, P. Clinical Relevance of Novel Polymorphisms in the Dihydropyrimidine Dehydrogenase (DPYD) Gene in Patients with Severe Fluoropyrimidine Toxicity: A Spanish Case-Control Study. Pharmaceutics 2021, 13, 2036. [Google Scholar] [CrossRef]
- Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Roselló, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized medicine: Recent progress in cancer therapy. Cancers 2020, 12, 1009. [Google Scholar] [CrossRef]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef]
- Koulis, C.; Yap, R.; Engel, R.; Jardé, T.; Wilkins, S.; Solon, G.; Shapiro, J.D.; Abud, H.; McMurrick, P. Personalized medicine—Current and emerging predictive and prognostic biomarkers in colorectal cancer. Cancers 2020, 12, 812. [Google Scholar] [CrossRef]
- Tyson, R.J.; Park, C.C.; Powell, J.R.; Patterson, J.H.; Weiner, D.; Watkins, P.B.; Gonzalez, D. Precision dosing priority criteria: Drug, disease, and patient population variables. Front. Pharmacol. 2020, 11, 420. [Google Scholar] [CrossRef]
- Singh, D.B. The impact of pharmacogenomics in personalized medicine. Curr. Appl. Pharm. Biotechnol. 2020, 45, 36–394. [Google Scholar]
- Micaglio, E.; Locati, E.T.; Monasky, M.M.; Pappone, C. Role of pharmacogenetics in adverse drug reactions: An update towards personalized medicine. Front. Pharmacol. 2021, 12, 651720. [Google Scholar] [CrossRef]
- Ara, I.; Maqbool, M.; Gani, I. Specificity and Personalized medicine: A novel approach to Cancer management. Int. J. Curr. Res. Physiol. Pharmacol. 2022, 6, 11–20. [Google Scholar]
- Ciardiello, F.; Ciardiello, D.; Martini, G.; Napolitano, S.; Tabernero, J.; Cervantes, A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J. Clin. 2022, 72, 372–401. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, C.; Zembutsu, H. Pharmacogenetics for severe adverse drug reactions induced by molecular-targeted therapy. Cancer Sci. 2020, 111, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.; De Bellis, E.; Manzo, V.; Sabbatino, F.; Iannello, F.; Dal Piaz, F.; Izzo, V.; Charlier, B.; Stefanelli, B.; Torsiello, M. A genotyping/phenotyping approach with careful clinical monitoring to manage the fluoropyrimidines-based therapy: Clinical cases and systematic review of the literature. J. Pers. Med. 2020, 10, 113. [Google Scholar] [CrossRef]
- Ridge, S.A.; Sludden, J.; Wei, X.; Sapone, A.; Brown, O.; Hardy, S.; Canney, P.; Fernandez-Salguero, P.; Gonzalez, F.J.; Cassidy, J.; et al. Dihydropyrimidine dehydrogenase pharmacogenetics in patients with colorectal cancer. Br. J. Cancer 1998, 77, 497–500. [Google Scholar] [CrossRef]
- Shrestha, S.; Zhang, C.; Jerde, C.R.; Nie, Q.; Li, H.; Offer, S.M.; Diasio, R.B. Gene-Specific Variant Classifier (DPYD-Varifier) to Identify Deleterious Alleles of Dihydropyrimidine Dehydrogenase. Clin. Pharmacol. Ther. 2018, 104, 709–718. [Google Scholar] [CrossRef]
- Chopra, D.; Rehan, H.S.; Sharma, V.; Mishra, R. Chemotherapy-induced adverse drug reactions in oncology patients: A prospective observational survey. Indian J. Med. Paediatr. Oncol. 2016, 37, 42–46. [Google Scholar] [CrossRef]
- Hurrell, T.; Naidoo, J.; Masimirembwa, C.; Scholefield, J. The Case for Pre-Emptive Pharmacogenetic Screening in South Africa. J. Pers. Med. 2024, 14, 114. [Google Scholar] [CrossRef] [PubMed]
- Knikman, J.E.; Wilting, T.A.; Lopez-Yurda, M.; Henricks, L.M.; Lunenburg, C.; de Man, F.M.; Meulendijks, D.; Nieboer, P.; Droogendijk, H.J.; Creemers, G.J.; et al. Survival of Patients With Cancer With DPYD Variant Alleles and Dose-Individualized Fluoropyrimidine Therapy-A Matched-Pair Analysis. J. Clin. Oncol. 2023, 41, 5411–5421. [Google Scholar] [CrossRef] [PubMed]
- Lanillos, J.; Carcajona, M.; Maietta, P.; Alvarez, S.; Rodriguez-Antona, C. Clinical pharmacogenetic analysis in 5,001 individuals with diagnostic Exome Sequencing data. NPJ Genom. Med. 2022, 7, 12. [Google Scholar] [CrossRef]
- Swen, J.J.; van der Wouden, C.H.; Manson, L.E.; Abdullah-Koolmees, H.; Blagec, K.; Blagus, T.; Bohringer, S.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; et al. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: An open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet 2023, 401, 347–356. [Google Scholar] [CrossRef] [PubMed]
| Characteristics | Cohort |
|---|---|
| Patients | 132 |
| Gender (M/F) | 72/60 |
| Age (years) | 66.5 ± 10.4 |
| Ancestry | Caucasian, European |
| Colorectal cancer | 91 (68.94%) |
| Gastric cancer | 37 (28.03%) |
| Breast cancer | 4 (3.03%) |
| Treatment | |
| FU-LV (De Gramont regimen) | 6 (4.5%) |
| 5-FU | 3 (2.3%) |
| FOLFOX | 90 (68.2%) |
| FOLFIRI | 14 (10.6%) |
| FOLFIRINOX | 15 (11.4%) |
| Capecitabine | 4 (3%) |
| Combination with immunotherapy | |
| Yes | 30 (22.7%) |
| No | 102 (77.3%) |
| Comorbidity | 34 (25.75%) |
| No Comorbidity | 98 (74.25%) |
| Side Effect | WT (n = 106) | DPYD*6 Polymorphism c.2194G>A (n = 26) | p-Value |
|---|---|---|---|
| ADRs—Gastrointestinal | |||
| Nausea/Vomiting (24 cases) | 14/106 | 10/26 | p = 0.002 |
| Diarrhea (56 cases) | 32/106 | 24/26 | p < 0.001 |
| Stomatitis (21 cases) | 15/106 | 6/26 | p = 0.03 |
| ADRs—Dermatological | |||
| Hand–foot syndrome (11 cases) | 8/106 | 3/26 | p = 0.45 |
| ADRs—Hematological | |||
| Fever (6 cases) | 3/106 | 3/26 | p = 0.09 |
| Leucopenia (19 cases) | 9/106 | 10/26 | p < 0.001 |
| Neutropenia (28 cases) | 16/106 | 12/26 | p = 0.002 |
| Anemia (7 cases) | 5/106 | 2/26 | p = 0.62 |
| Thrombocytopenia (9 cases) | 4/106 | 5/26 | p = 0.01 |
| ADRs—Neurological | |||
| Peripheral Neuropathy (4 cases) | 2/106 | 2/26 | p = 0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ardizzone, A.; Bulzomì, M.; De Luca, F.; Silvestris, N.; Esposito, E.; Capra, A.P. Dihydropyrimidine Dehydrogenase Polymorphism c.2194G>A Screening Is a Useful Tool for Decreasing Gastrointestinal and Hematological Adverse Drug Reaction Risk in Fluoropyrimidine-Treated Patients. Curr. Issues Mol. Biol. 2024, 46, 9831-9843. https://doi.org/10.3390/cimb46090584
Ardizzone A, Bulzomì M, De Luca F, Silvestris N, Esposito E, Capra AP. Dihydropyrimidine Dehydrogenase Polymorphism c.2194G>A Screening Is a Useful Tool for Decreasing Gastrointestinal and Hematological Adverse Drug Reaction Risk in Fluoropyrimidine-Treated Patients. Current Issues in Molecular Biology. 2024; 46(9):9831-9843. https://doi.org/10.3390/cimb46090584
Chicago/Turabian StyleArdizzone, Alessio, Maria Bulzomì, Fabiola De Luca, Nicola Silvestris, Emanuela Esposito, and Anna Paola Capra. 2024. "Dihydropyrimidine Dehydrogenase Polymorphism c.2194G>A Screening Is a Useful Tool for Decreasing Gastrointestinal and Hematological Adverse Drug Reaction Risk in Fluoropyrimidine-Treated Patients" Current Issues in Molecular Biology 46, no. 9: 9831-9843. https://doi.org/10.3390/cimb46090584