_Kim.png)
Identification of Potential Selective PAK4 Inhibitors Through Shape and Protein Conformation Ensemble Screening and Electrostatic-Surface-Matching Optimization



Abstract
1. Introduction
2. Materials and Methods
2.1. Protein and Ligand Preparation
2.2. Cross-Docking
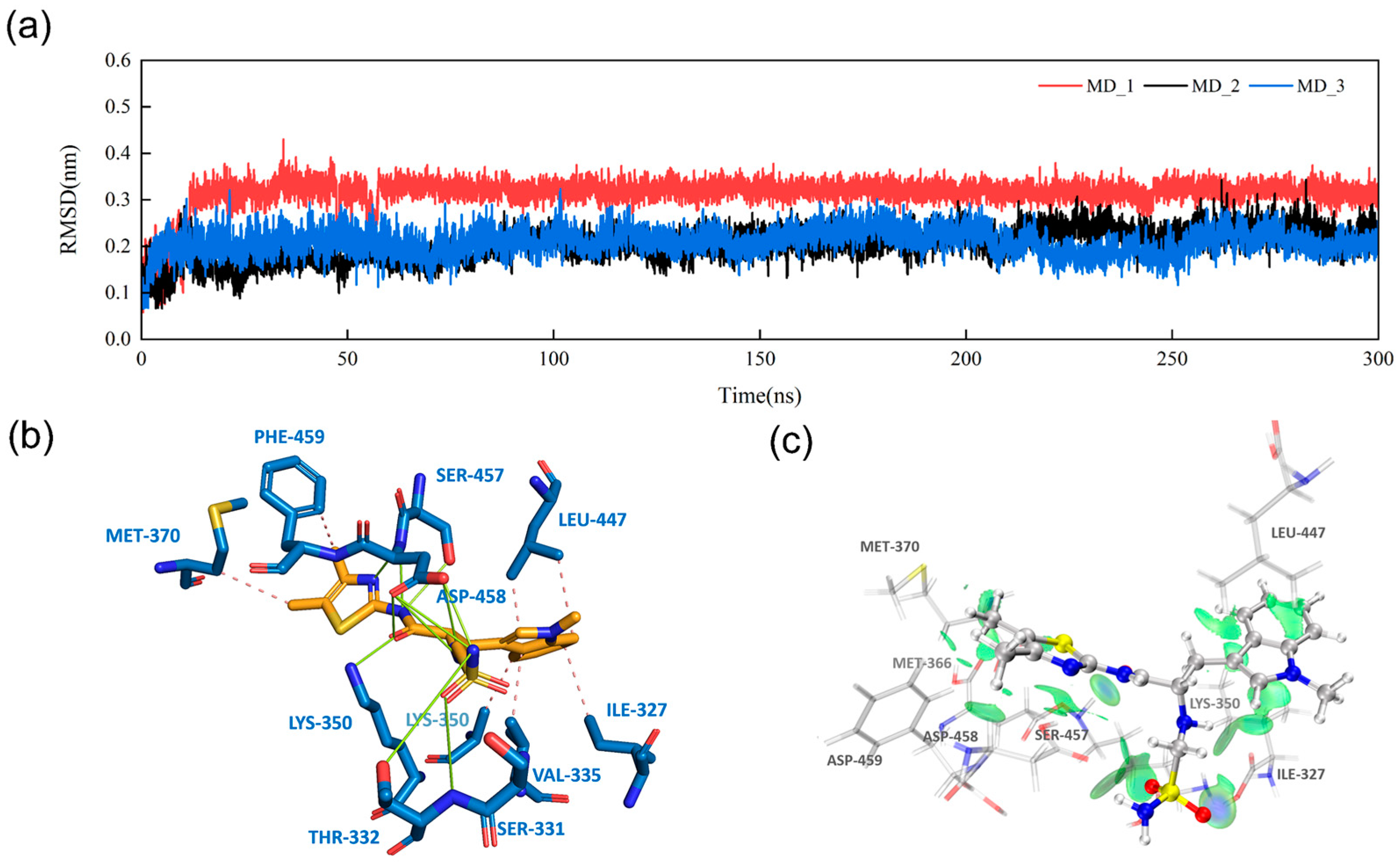
2.3. Unbiased Molecular Dynamics Simulations
2.4. Binding Energy Calculation
2.5. Shape Screening
2.6. Virtual Screening Based on Protein Conformational Ensembles
2.7. Re-Scoring of Docking Conformation
2.8. Molecular Optimization and Analysis
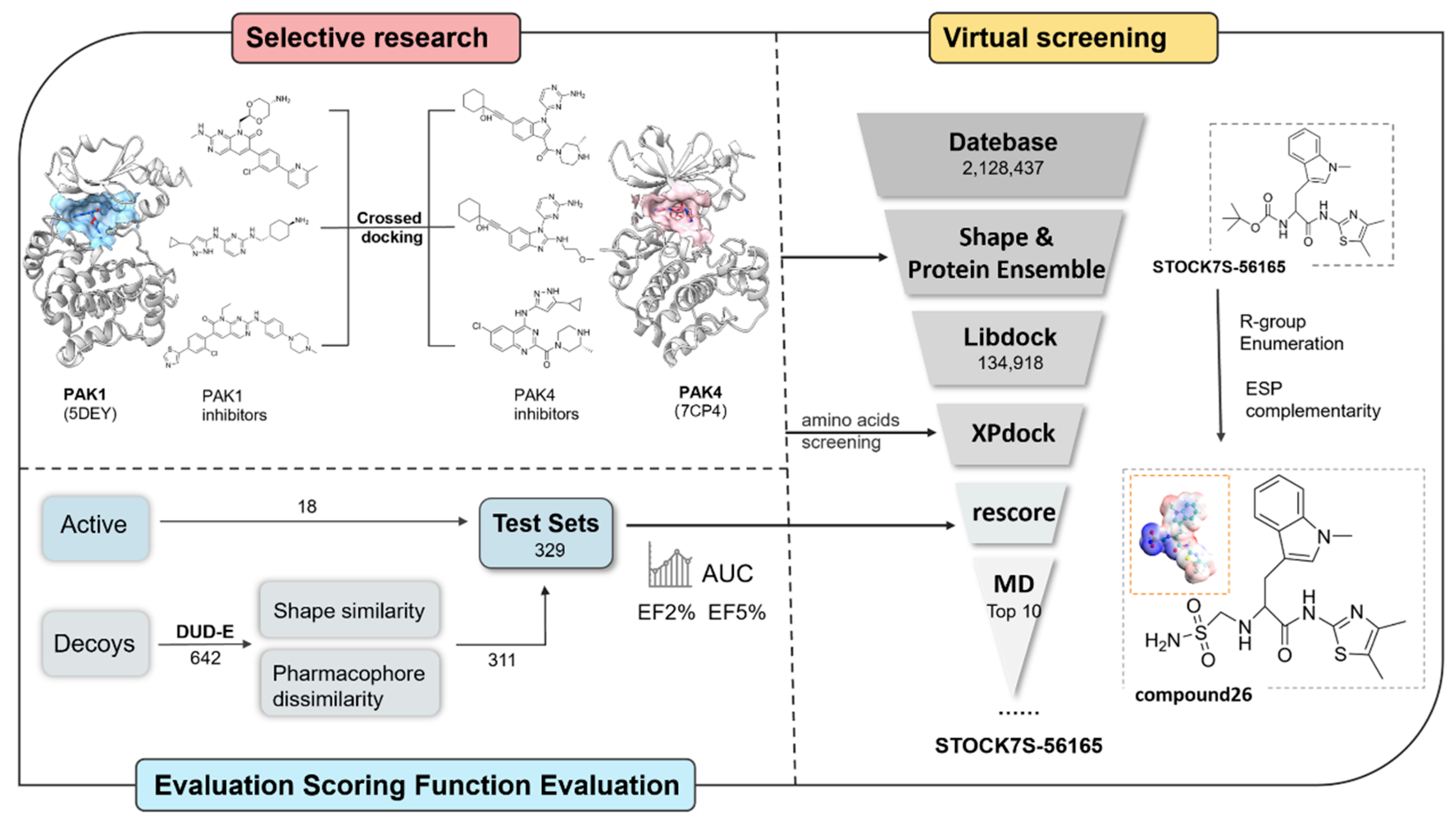
3. Results
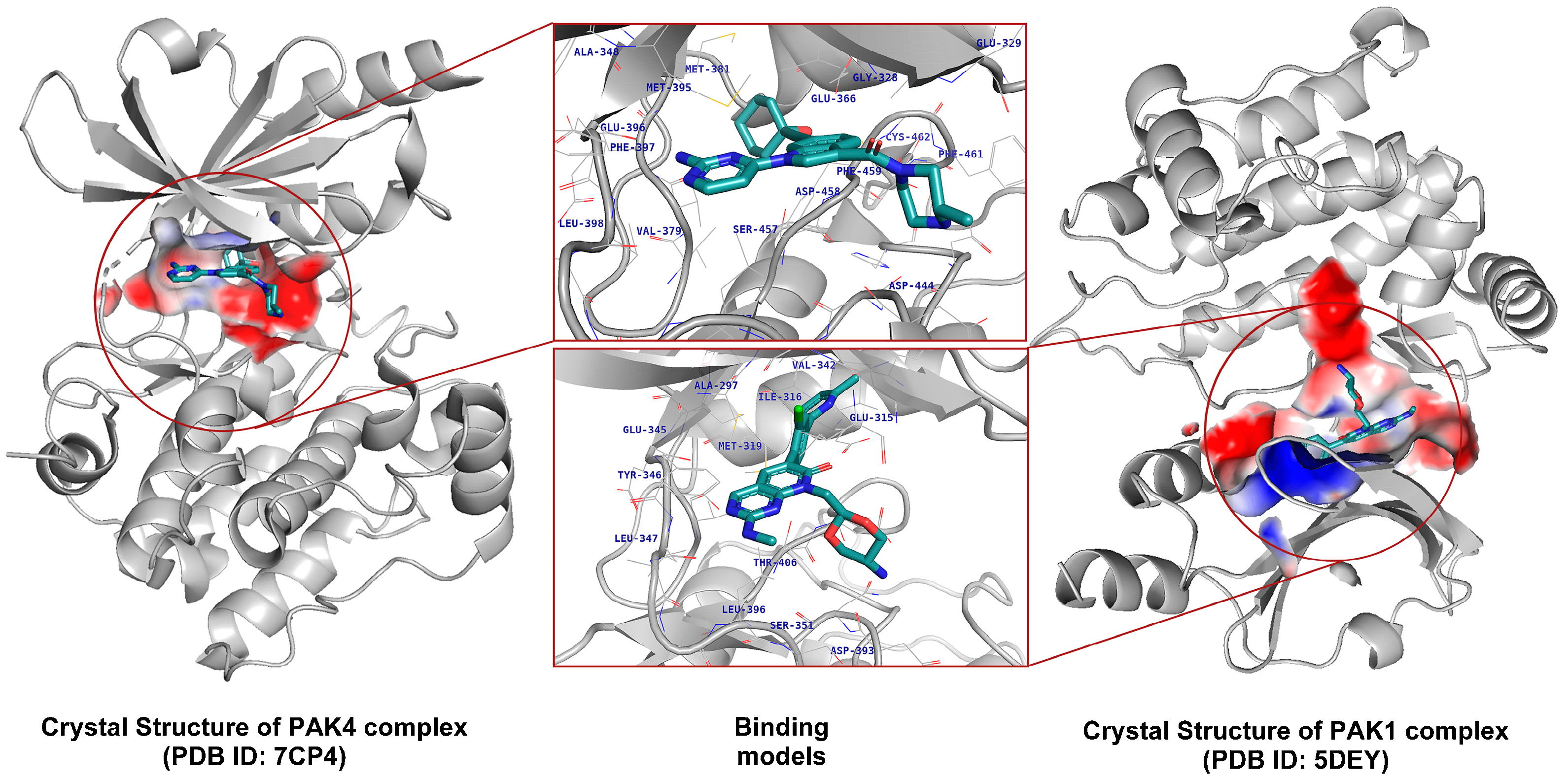
3.1. Structural Comparison and Binding Site Analysis of PAK1 and PAK4
3.2. Virtual Screening Based on Shape and Protein Conformational Ensembles
3.3. Re-Scoring to Improve Screening Performance
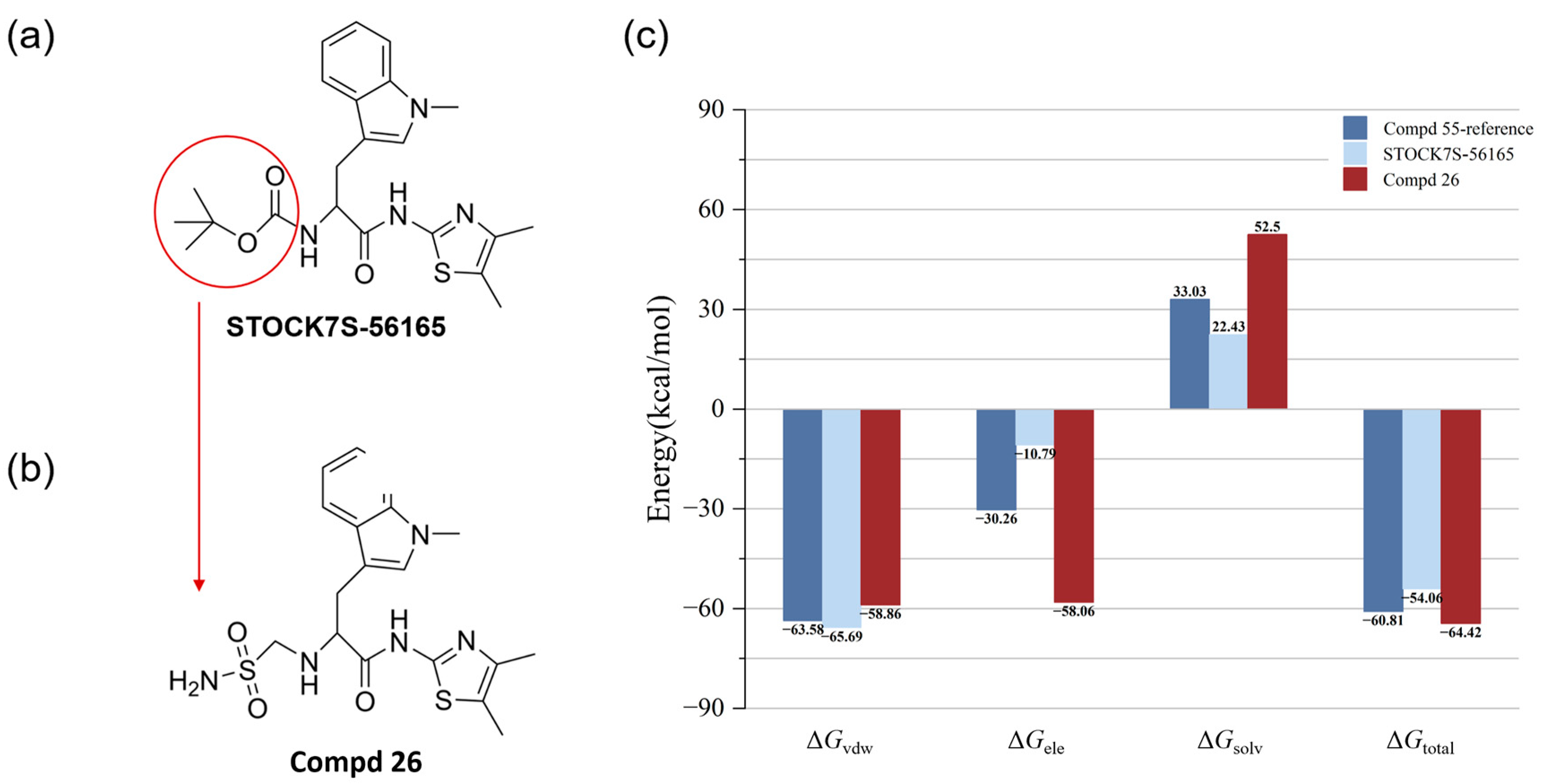
3.4. Optimization of Candidate Molecules by Electrostatic Surface Matching Combined with Fragment Substitution
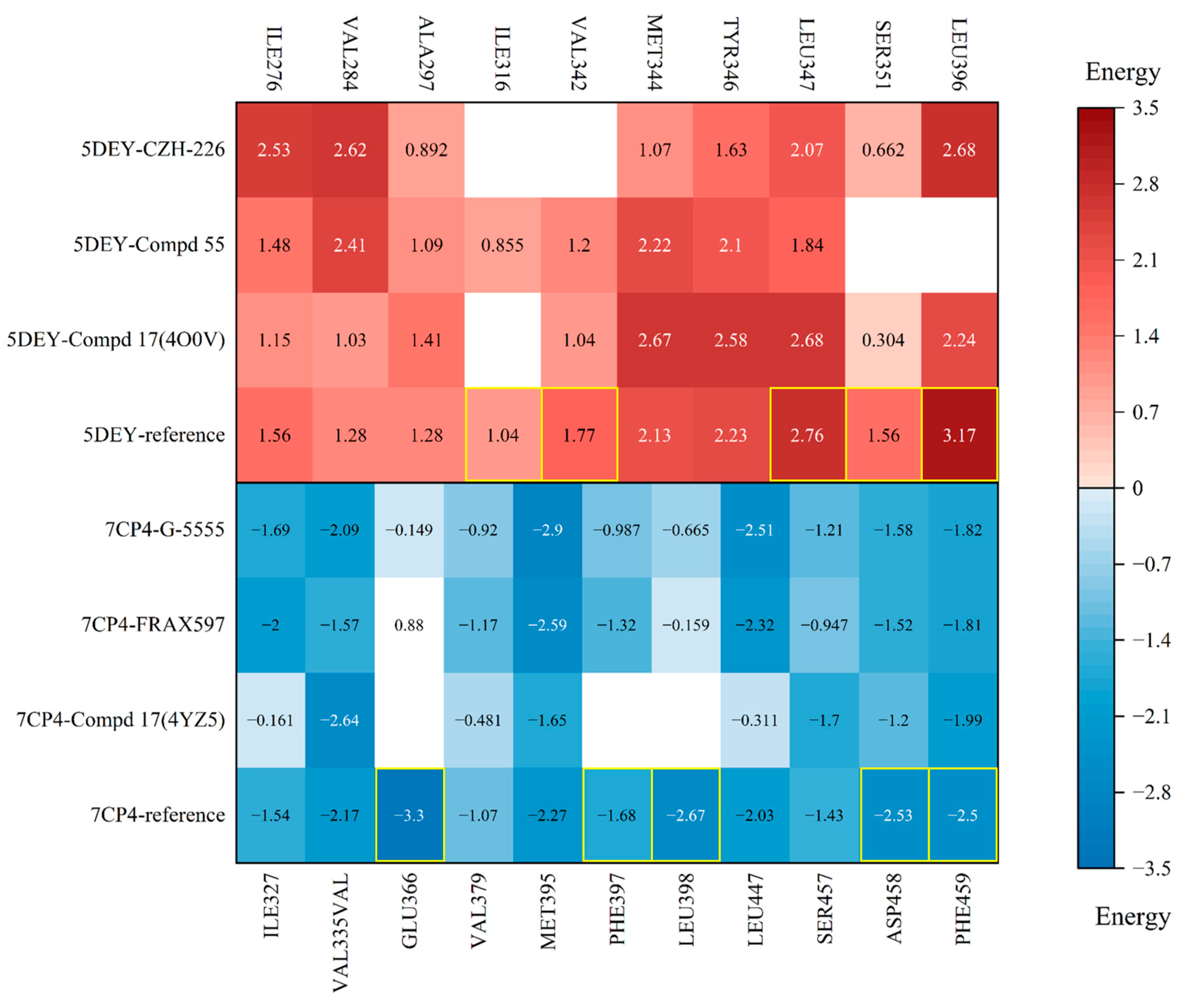
3.5. Intermolecular Interactions and IGMH Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molli, P.R.; Li, D.Q.; Murray, B.W.; Rayala, S.K.; Kumar, R. PAK Signaling in Oncogenesis. Oncogene 2009, 28, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Bagrodia, S.; Cerione, R.A. Pak to the Future. Trends Cell Biol. 1999, 9, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Q.; Elnitski, L. A Systems Biology Comparison of Ovarian Cancers Implicates Putative Somatic Driver Mutations through Protein-Protein Interaction Models. PLoS ONE 2016, 11, e0163353. [Google Scholar] [CrossRef] [PubMed]
- King, H.; Thillai, K.; Whale, A.; Arumugam, P.; Eldaly, H.; Kocher, H.M.; Wells, C.M. PAK4 Interacts with P85 Alpha: Implications for Pancreatic Cancer Cell Migration. Sci. Rep. 2017, 7, 42575. [Google Scholar] [CrossRef]
- Rane, C.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Crochiere, M.; Das-Gupta, S.; Minden, A. A Novel Orally Bioavailable Compound KPT-9274 Inhibits PAK4, and Blocks Triple Negative Breast Cancer Tumor Growth. Sci. Rep. 2017, 7, 42555. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Faruqu, F.N.; Lim, Y.M.; Lim, K.Y.; Liam-Or, R.; Walters, A.A.; Lavender, P.; Fear, D.; Wells, C.M.; Tzu-Wen Wang, J.; et al. Exosome-Mediated RNAi of PAK4 Prolongs Survival of Pancreatic Cancer Mouse Model after Loco-Regional Treatment. Biomaterials 2021, 264, 120369. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Ye, Z.; Wang, X.; Pan, Y.; Weng, Y.; Lao, S.; Wei, H.; Li, L. Overexpression of P21-Activated Kinase 4 Is Associated with Poor Prognosis in Non-Small Cell Lung Cancer and Promotes Migration and Invasion. J. Exp. Clin. Cancer Res. 2015, 34, 48. [Google Scholar] [CrossRef]
- Wang, M.; Gao, Q.; Chen, Y.; Li, Z.; Yue, L.; Cao, Y. PAK4, a Target of miR-9-5p, Promotes Cell Proliferation and Inhibits Apoptosis in Colorectal Cancer. Cell. Mol. Biol. Lett. 2019, 24, 58. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Du, R.; Jia, X.; Liu, K.; Qiao, Y.; Wu, Q.; Yao, N.; Yang, L.; Zhou, L.; Liu, X.; et al. CDK15 Promotes Colorectal Cancer Progression via Phosphorylating PAK4 and Regulating β-Catenin/ MEK-ERK Signaling Pathway. Cell Death Differ. 2022, 29, 14–27. [Google Scholar] [CrossRef]
- Liu, Y.; Xiao, H.; Tian, Y.; Nekrasova, T.; Hao, X.; Lee, H.J.; Suh, N.; Yang, C.S.; Minden, A. The Pak4 Protein Kinase Plays a Key Role in Cell Survival and Tumorigenesis in Athymic Mice. Mol. Cancer Res. 2008, 6, 1215–1224. [Google Scholar] [CrossRef]
- Wang, H.; Song, P.; Gao, Y.; Shen, L.; Xu, H.; Wang, J.; Cheng, M. Drug Discovery Targeting P21-Activated Kinase 4 (PAK4): A Patent Review. Expert Opin. Ther. Pat. 2021, 31, 977–987. [Google Scholar] [CrossRef]
- Yu, X.; Huang, C.; Liu, J.; Shi, X.; Li, X. The Significance of PAK4 in Signaling and Clinicopathology: A Review. Open Life Sci. 2022, 17, 586–598. [Google Scholar] [CrossRef]
- Cordover, E.; Wei, J.; Patel, C.; Shan, N.L.; Gionco, J.; Sargsyan, D.; Wu, R.; Cai, L.; Kong, A.-N.; Jacinto, E.; et al. KPT-9274, an Inhibitor of PAK4 and NAMPT, Leads to Downregulation of mTORC2 in Triple Negative Breast Cancer Cells. Chem. Res. Toxicol. 2020, 33, 482–491. [Google Scholar] [CrossRef]
- Abril-Rodriguez, G.; Torrejon, D.Y.; Liu, W.; Zaretsky, J.M.; Nowicki, T.S.; Tsoi, J.; Puig-Saus, C.; Baselga-Carretero, I.; Medina, E.; Quist, M.J.; et al. PAK4 Inhibition Improves PD-1 Blockade Immunotherapy. Nat. Cancer 2019, 1, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Fessler, J. PAK4 as a Cancer Immune-Evasion Target. Nat. Cancer 2020, 1, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.W.; Guo, C.; Piraino, J.; Westwick, J.K.; Zhang, C.; Lamerdin, J.; Dagostino, E.; Knighton, D.; Loi, C.-M.; Zager, M.; et al. Small-Molecule P21-Activated Kinase Inhibitor PF-3758309 Is a Potent Inhibitor of Oncogenic Signaling and Tumor Growth. Proc. Natl. Acad. Sci. USA 2010, 107, 9446–9451. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Huang, W.; Li, X.; Guo, J.; Chen, M.; Yan, Z.; Wang, K.; Jiang, X.; Song, S.; Wang, J.; et al. Development of 2, 4-Diaminoquinazoline Derivatives as Potent PAK4 Inhibitors by the Core Refinement Strategy. Eur. J. Med. Chem. 2017, 131, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ryu, B.J.; Kim, S.; Min, B.; Kim, K.Y.; Lee, J.S.; Park, W.J.; Lee, H.; Kim, S.H.; Park, S. Discovery and the Structural Basis of a Novel P21-Activated Kinase 4 Inhibitor. Cancer Lett. 2014, 349, 45–50. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Guo, Q.; Wang, Y.; Zhou, Y.; Peng, H.; Cheng, M.; Zhao, D.; Li, F. LCH-7749944, a Novel and Potent P21-Activated Kinase 4 Inhibitor, Suppresses Proliferation and Invasion in Human Gastric Cancer Cells. Cancer Lett. 2012, 317, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. Schrödinger Release 2022-3; Schrödinger, LLC: New York, NY, USA, 2022. [Google Scholar]
- Staben, S.T.; Feng, J.A.; Lyle, K.; Belvin, M.; Boggs, J.; Burch, J.D.; Chua, C.; Cui, H.; DiPasquale, A.G.; Friedman, L.S.; et al. Back Pocket Flexibility Provides Group II P21-Activated Kinase (PAK) Selectivity for Type I 1/2 Kinase Inhibitors. J. Med. Chem. 2014, 57, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zhao, F.; Li, D.; Qu, J.; Yao, M.; Su, Y.; Wang, H.; Zhou, M.; Wang, Y.; Gao, Y.; et al. Synthesis of Selective PAK4 Inhibitors for Lung Metastasis of Lung Cancer and Melanoma Cells. Acta Pharm. Sin. B 2022, 12, 2905–2922. [Google Scholar] [CrossRef]
- Ndubaku, C.O.; Crawford, J.J.; Drobnick, J.; Aliagas, I.; Campbell, D.; Dong, P.; Dornan, L.M.; Duron, S.; Epler, J.; Gazzard, L.; et al. Design of Selective PAK1 Inhibitor G-5555: Improving Properties by Employing an Unorthodox Low-p K a Polar Moiety. ACS Med. Chem. Lett. 2015, 6, 1241–1246. [Google Scholar] [CrossRef]
- Licciulli, S.; Maksimoska, J.; Zhou, C.; Troutman, S.; Kota, S.; Liu, Q.; Duron, S.; Campbell, D.; Chernoff, J.; Field, J.; et al. FRAX597, a Small Molecule Inhibitor of the P21-Activated Kinases, Inhibits Tumorigenesis of Neurofibromatosis Type 2 (NF2)-Associated Schwannomas. J. Biol. Chem. 2013, 288, 29105–29114. [Google Scholar] [CrossRef]
- Crawford, J.J.; Lee, W.; Aliagas, I.; Mathieu, S.; Hoeflich, K.P.; Zhou, W.; Wang, W.; Rouge, L.; Murray, L.; La, H.; et al. Structure-Guided Design of Group I Selective P21-Activated Kinase Inhibitors. J. Med. Chem. 2015, 58, 5121–5136. [Google Scholar] [CrossRef]
- Hao, C.; Zhao, F.; Song, H.; Guo, J.; Li, X.; Jiang, X.; Huan, R.; Song, S.; Zhang, Q.; Wang, R.; et al. Structure-Based Design of 6-Chloro-4-Aminoquinazoline-2-Carboxamide Derivatives as Potent and Selective P21-Activated Kinase 4 (PAK4) Inhibitors. J. Med. Chem. 2018, 61, 265–285. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.O1; Gaussian: Wallingford, CT, USA, 2013. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force fiel. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Tian, L. Sobtop, Version 1.0 (dev3.1). Available online: http://sobereva.com/soft/Sobtop/ (accessed on 9 August 2024).
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.I.; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Bento, A.P.; Hersey, A.; Félix, E.; Landrum, G.; Gaulton, A.; Atkinson, F.; Bellis, L.J.; De Veij, M.; Leach, A.R. An Open Source Chemical Structure Curation Pipeline Using RDKit. J. Cheminform. 2020, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking\textbar Journal of Medicinal Chemistry. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of Machine-Learning Scoring Functions in Structure-Based Virtual Screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A New Open-Source Player in the Drug Discovery Field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mitchell, J.B. Machine Learning Approach to Predicting Protein–Ligand Binding Affinity with Applications to Molecular Docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef]
- Ballester, P.J.; Schreyer, A.; Blundell, T.L. Does a More Precise Chemical Description of Protein–Ligand Complexes Lead to More Accurate Prediction of Binding Affinity? J. Chem. Inf. Model. 2014, 54, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leung, K.S.; Wong, M.H.; Ballester, P.J. Improving AutoDock Vina Using Random Forest: The Growing Accuracy of Binding Affinity Prediction by the Effective Exploitation of Larger Data Sets. Mol. Inform. 2015, 34, 115–126. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular Docking with Deep Learning. J. Cheminform. 2021, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q. Independent Gradient Model Based on Hirshfeld Partition: A New Method for Visual Study of Interactions in Chemical Systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Distinto, S.; Markt, P.; Schuster, D.; Spitzer, G.M.; Liedl, K.R.; Wolber, G. How to Optimize Shape-Based Virtual Screening: Choosing the Right Query and Including Chemical Information. J. Chem. Inf. Model. 2009, 49, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.J.; Lamb, M.L.; Joseph-McCarthy, D. Ensemble-Based Docking Using Biased Molecular Dynamics. J. Chem. Inf. Model. 2014, 54, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Bietz, S.; Rarey, M. SIENA: Efficient Compilation of Selective Protein Binding Site Ensembles. J. Chem. Inf. Model. 2016, 56, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Jiang, K.; Teng, D.; Wu, Z.; Li, W.; Tang, Y.; Wang, R.; Liu, G. Discovery of New Estrogen-Related Receptor α Agonists via a Combination Strategy Based on Shape Screening and Ensemble Docking. J. Chem. Inf. Model. 2022, 62, 486–497. [Google Scholar] [CrossRef]
- Yang, J.; Baek, M.; Seok, C. GalaxyDock3: Protein–Ligand Docking That Considers the Full Ligand Conformational Flexibility. J. Comput. Chem. 2019, 40, 2739–2748. [Google Scholar] [CrossRef]
- Abdel-Rahman, S.A.; Ovchinnikov, V.; Gabr, M.T. Structure-Based Rational Design of Constrained Peptides as TIM-3 Inhibitors. ACS Med. Chem. Lett. 2024, 15, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Tran-Nguyen, V.-K.; Bret, G.; Rognan, D. True Accuracy of Fast Scoring Functions to Predict High-Throughput Screening Data from Docking Poses: The Simpler the Better. J. Chem. Inf. Model. 2021, 61, 2788–2797. [Google Scholar] [CrossRef] [PubMed]
- Scantlebury, J.; Vost, L.; Carbery, A.; Hadfield, T.E.; Turnbull, O.M.; Brown, N.; Chenthamarakshan, V.; Das, P.; Grosjean, H.; von Delft, F.; et al. A Small Step Toward Generalizability: Training a Machine Learning Scoring Function for Structure-Based Virtual Screening. J. Chem. Inf. Model. 2023, 63, 2960–2974. [Google Scholar] [CrossRef] [PubMed]
- Cons, B.D.; Twigg, D.G.; Kumar, R.; Chessari, G. Electrostatic Complementarity in Structure-Based Drug Design. J. Med. Chem. 2022, 65, 7476–7488. [Google Scholar] [CrossRef] [PubMed]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the Scope of the Protein–Ligand Interaction Profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EF2% | EF5% | AUC | |
|---|---|---|---|
| RF-VS | 10.45 | 8.00 | 0.86 |
| CNNaffinity | 18.28 | 7.80 | 0.83 |
| CNNscore | 15.67 | 9.14 | 0.71 |
| Affinity | 10.45 | 4.57 | 0.80 |
| CNN_VS | 18.28 | 8.00 | 0.72 |
| DockingScore | 15.67 | 8.00 | 0.82 |
| XPscore | 15.67 | 9.14 | 0.83 |
| GlideScore | 15.67 | 8.00 | 0.83 |
| Lig1 | 7.83 | 4.57 | 0.80 |
| Lig2 | 15.67 | 10.28 | 0.87 |
| PLP1 | 15.67 | 7.80 | 0.86 |
| PLP2 | 15.67 | 9.14 | 0.86 |
| Jain | 10.45 | 5.71 | 0.85 |
| PMF | 13.06 | 8.00 | 0.87 |
| PMF04 | 0 | 1.14 | 0.72 |
| Compounds | MM/GBSA | GNINA (CNN_VS) | |||
|---|---|---|---|---|---|
| ΔGvdw | ΔGele | ΔGGB | ΔGtotal | ||
| Compd 55 | −63.58 | −30.26 | 39.99 | −60.81 | 7.61 |
| HIT213882013 | −49.18 | −19.88 | 29.30 | −46.07 | 7.50 |
| STOCK1S-85434 | −66.13 | −18.27 | 40.60 | −51.98 | 7.63 |
| HIT104079502 | −51.46 | −18.26 | 35.34 | −40.69 | 7.60 |
| SN0341269 | −52.33 | −17.66 | 42.92 | −34.12 | 7.60 |
| STOCK7S-56165 | −65.69 | −10.79 | 29.69 | −54.06 | 7.58 |
| HIT212577525 | −53.27 | −38.81 | 47.34 | −51.10 | 7.47 |
| HIT105326727 | −51.16 | −38.85 | 55.42 | −41.80 | 7.46 |
| HIT105409527 | −45.53 | −28.69 | 46.82 | −33.59 | 7.41 |
| HIT213881679 | −58.67 | −40.38 | 58.04 | −47.43 | 7.36 |
| HIT104998753 | −52.48 | −32.36 | 43.86 | −47.41 | 7.35 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Zhang, M.; Li, Y.; Deng, P. Identification of Potential Selective PAK4 Inhibitors Through Shape and Protein Conformation Ensemble Screening and Electrostatic-Surface-Matching Optimization. Curr. Issues Mol. Biol. 2025, 47, 29. https://doi.org/10.3390/cimb47010029
Zhang X, Zhang M, Li Y, Deng P. Identification of Potential Selective PAK4 Inhibitors Through Shape and Protein Conformation Ensemble Screening and Electrostatic-Surface-Matching Optimization. Current Issues in Molecular Biology. 2025; 47(1):29. https://doi.org/10.3390/cimb47010029
Chicago/Turabian StyleZhang, Xiaoxuan, Meile Zhang, Yihao Li, and Ping Deng. 2025. "Identification of Potential Selective PAK4 Inhibitors Through Shape and Protein Conformation Ensemble Screening and Electrostatic-Surface-Matching Optimization" Current Issues in Molecular Biology 47, no. 1: 29. https://doi.org/10.3390/cimb47010029
APA StyleZhang, X., Zhang, M., Li, Y., & Deng, P. (2025). Identification of Potential Selective PAK4 Inhibitors Through Shape and Protein Conformation Ensemble Screening and Electrostatic-Surface-Matching Optimization. Current Issues in Molecular Biology, 47(1), 29. https://doi.org/10.3390/cimb47010029