_Kim.png)
The Role of the Arcuate Nucleus in Regulating Hunger and Satiety in Prader-Willi Syndrome


Abstract
1. Introduction
2. The Role of the Hypothalamus in Appetite and Metabolism
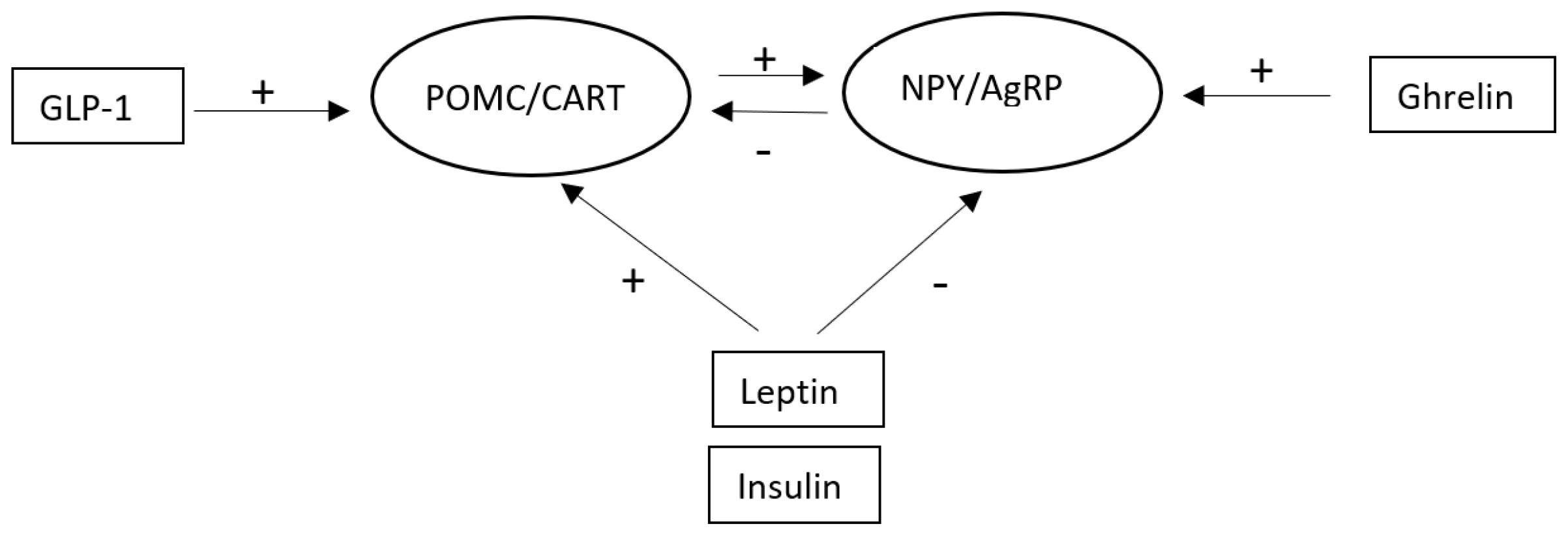
2.1. The Role of the Arcuate Nucleus in Appetite and Metabolism
2.2. POMC/CART Neurons
2.3. NPY/AgRP Neurons
2.4. Other Neurons in the Arcuate Nucleus
3. The Arcuate Nucleus in PWS
4. How Peptides and Proteins Produced Within the Brain Affect the Arcuate Nucleus—And Their Potential Roles in PWS
4.1. Oxytocin
4.2. Brain-Derived Neurotrophic Factor
4.3. Orexin
4.4. Kisspeptin
4.5. Tachykinins
4.6. Nesfatin-1
4.7. CRH
4.8. TRH
4.9. GHRH
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Driscoll, D.J.; Miller, J.L.; Cassidy, S.B. Prader-Willi Syndrome; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; GeneReviews®: Seattle, WA, USA; University of Washington: Seattle, WA, USA, 1998; pp. 1993–2024, [updated 5 December 2024]. [Google Scholar] [PubMed]
- Bar, C.; Diene, G.; Molinas, C.; Bieth, E.; Casper, C.; Tauber, M. Early diagnosis and care is achieved but should be improved in infants with Prader-Willi syndrome. Orphanet J. Rare Dis. 2017, 12, 118. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tauber, M.; Hoybye, C. Endocrine disorders in Prader-Willi syndrome: A model to understand and treat hypothalamic dysfunction. Lancet Diabetes Endocrinol. 2021, 9, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Correa-da-Silva, F.; Fliers, E.; Swaab, D.F.; Yi, C.X. Hypothalamic neuropeptides and neurocircuitries in Prader Willi syndrome. J. Neuroendocrinol. 2021, 33, e12994. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bochukova, E.G.; Lawler, K.; Croizier, S.; Keogh, J.M.; Patel, N.; Strohbehn, G.; Lo, K.K.; Humphrey, J.; Hokken-Koelega, A.; Damen, L.; et al. A Transcriptomic Signature of the Hypothalamic Response to Fasting and BDNF Deficiency in Prader-Willi Syndrome. Cell Rep. 2018, 22, 3401–3408. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brown, S.S.G.; Manning, K.E.; Fletcher, P.; Holland, A. In vivo neuroimaging evidence of hypothalamic alteration in Prader-Willi syndrome. Brain Commun. 2022, 4, fcac229. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, N.; Yu, H.; Xu, M. Alteration of brain nuclei in obese children with and without Prader-Willi syndrome. Front. Neuroinform. 2022, 16, 1032636. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Holsen, L.M.; Zarcone, J.R.; Brooks, W.M.; Butler, M.G.; Thompson, T.I.; Ahluwalia, J.S.; Nollen, N.L.; Savage, C.R. Neural mechanisms underlying hyperphagia in Prader–Willi syndrome. Obesity 2006, 14, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Holsen, L.M.; Savage, C.R.; Martin, L.E.; Bruce, A.S.; Lepping, R.J.; Ko, E.; Brooks, W.M.; Butler, M.G.; Zarcone, J.R.; Goldstein, J.M. Importance of reward and prefrontal circuitry in hunger and satiety: Prader–Willi syndrome vs simple obesity. Int. J. Obes. 2012, 36, 638–647. [Google Scholar] [CrossRef]
- Akefeldt, A.; Ekman, R.; Gillberg, C.; Månsson, J.E. Cerebrospinal fluid monoamines in Prader-Willi syndrome. Biol. Psychiatry 1998, 44, 1321–1328. [Google Scholar] [CrossRef]
- Song, J.; Choi, S.Y. Arcuate Nucleus of the Hypothalamus: Anatomy, Physiology, and Diseases. Exp. Neurobiol. 2023, 32, 371–386. [Google Scholar] [CrossRef]
- Vohra, M.S.; Benchoula, K.; Serpell, C.J.; Hwa, W.E. AgRP/NPY and POMC neurons in the arcuate nucleus and their potential role in treatment of obesity. Eur. J. Pharmacol. 2022, 915, 174611. [Google Scholar] [CrossRef]
- Crino, A.; Graziano, G. Updated on diabetes mellitus and glucose metabolism alterations in Prader-Willi Syndrome. Curr. Diab Rep. 2020, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, K.A.; Bain, J.; Butler, M.G.; Ilkayeva, O.; Muehlbauer, M.; Haqq, A.M.; Freemark, M. Metabolic profiling in Prader-Willi syndrome and nonsyndromic obesity: Sex differences and the role of growth hormone. Clin. Endocrinol. 2015, 83, 797–805. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aponte, Y.; Atasoy, D.; Sternson, S.M. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci. 2011, 14, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.A. Stress, Neuropeptides, and Feeding Behavior: A Comparative Perspective. Integr. Comp. Biol. 2002, 42, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.E.; Grove, K.L.; Smith, M.S. Distribution of corticotropin releasing hormone receptor immunoreactivity in the rat hypothalamus: Coexpression in neuropeptide Y and dopamine neurons in the arcuate nucleus. Brain Res. 2003, 973, 223–232. [Google Scholar] [CrossRef]
- Marie, L.S.; Luquet, S.; Cole, T.B.; Palmiter, R.D. Modulation of neuropeptide Y expression in adult mice does not affect feeding. Proc. Natl. Acad. Sci. USA 2005, 102, 18632–18637. [Google Scholar] [CrossRef]
- Naveilhan, P.; Hassani, H.; Canals, J.M.; Ekstrand, A.J.; Larfalk, A.; Chhajlani, V.; Arenas, E.; Gedda, K.; Svensson, L.; Thoren, P.; et al. Normal feeding behavior, body weight and leptin response require the neuropeptide Y Y2 receptor. Nat. Med. 1999, 5, 1188–1193. [Google Scholar] [CrossRef]
- Qian, S.; Chen, H.; Weingarth, D.; Trumbauer, M.E.; Novi, D.E.; Guan, X.; Yu, H.; Shen, Z.; Feng, Y.; Frazier, E.; et al. Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol. Cell Biol. 2002, 22, 5027–5035. [Google Scholar] [CrossRef]
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 1997, 278, 135–138. [Google Scholar] [CrossRef]
- Bervini, S.; Herzog, H. Mouse models of Prader-Willi Syndrome: A systematic review. Front. Neuroendocrinol. 2013, 34, 107–119. [Google Scholar] [CrossRef]
- Choi, Y.; Min, H.Y.; Hwang, J.; Jo, Y.H. Magel2 knockdown in hypothalamic POMC neurons innervating the medial amygdala reduces susceptibility to diet-induced obesity. Life Sci. Alliance 2022, 5, e202201502. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wijesuriya, T.M.; De Ceuninck, L.; Masschaele, D.; Sanderson, M.R.; Carias, K.V.; Tavernier, J.; Wevrick, R. The Prader-Willi syndrome proteins MAGEL2 and necdin regulate leptin receptor cell surface abundance through ubiquitination pathways. Hum. Mol. Genet. 2017, 26, 4215–4230. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pravdivyi, I.; Ballanyi, K.; Colmers, W.F.; Wevrick, R. Progressive postnatal decline in leptin sensitivity of arcuate hypothalamic neurons in the Magel2-null mouse model of Prader-Willi syndrome. Hum. Mol. Genet. 2015, 24, 4276–4283. [Google Scholar] [CrossRef]
- Buch, J.R.; Wevrick, R. Loss of the Prader-Willi obesity syndrome protein necdin promotes adipogeneis. Gene 2021, 497, 45–51. [Google Scholar]
- Polex-Wolf, J.; Lam, B.Y.H.; Larder, R.; Tadross, J.; Rimmington, D.; Bosch, F.; Cenzano, V.J.; Eduard Ayuso, E.; Ma, M.K.L.; Rainbow, K.; et al. Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi Syndrome. J. Clin. Investig. 2018, 128, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Carraro, R.S.; Nogueira, G.A.; Sidarta-Oliveira, D.; Gaspar, R.S.; Dragano, N.R.; Morari, J.; Bobbo, V.C.D.; Araujo, E.P.; Mendes, N.F.; Zanesco, A.M.; et al. Arcuate Nucleus Overexpression of NHLH2 Reduces Body Mass and Attenuates Obesity-Associated Anxiety/Depression-like Behavior. J. Neurosci. 2021, 41, 10004–10022. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Topaloglu, A.K.; Simsek, E.; Kocher, M.A.; Mammadova, J.; Bober, E.; Kotan, L.D.; Turan, I.; Celiloglu, C.; Gurbuz, F.; Yuksel, B.; et al. Inactivating NHLH2 variants cause idiopathic hypogonadotropic hypogonadism and obesity in humans. Hum. Genet. 2022, 141, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Ohta, T.; Driscoll, D.J.; Nicholls, R.D.; Kalra, S.P. Anorexigenic melanocortin signaling in the hypothalamus is augmented in association with failure-to-thrive in a transgenic mouse model for Prader-Willi syndrome. Brain Res. 2002, 957, 42–45. [Google Scholar] [CrossRef]
- Goldstone, A.P.; Unmehopa, U.A.; Bloom, S.R.; Swaab, D.F. Hypothalamic NPY and agouti-related protein are increased in human illness but not in Prader-Willi syndrome and other obese subjects. J. Clin. Endocrinol. Metab. 2002, 87, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Turkkahraman, D.; Sirazi, E.C.; Aykal, G. Serum alpha-melanocyte-stimulating hormone (α-MSH), brain-derived neurotrophic factor (BDNF), and agouti-related protein (AGRP) levels in children with Prader-Willi or Bardet-Biedl syndromes. J. Endocrinol. Investig. 2022, 45, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- DelParigi, A.; Tschöp, M.; Heiman, M.L.; Salbe, A.D.; Vozarova, B.; Sell, S.M.; Bunt, J.C.; Tataranni, P.A. High circulating ghrelin: A potential cause for hyperphagia and obesity in Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2002, 87, 5461–5464. [Google Scholar] [CrossRef]
- Ding, F.; Li, H.H.; Zhang, S.; Solomon, N.M.; Camper, S.A.; Cohen, P.; Francke, U. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE 2008, 3, e1709. [Google Scholar] [CrossRef]
- Höybye, C.; Barkeling, B.; Espelund, U.; Petersson, M.; Thoren, M. Peptides associated with hyperphagia in adults with Prader–Willi syndrome before and during GH treatment. Growth Horm. IGF Res. 2003, 13, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Spetter, M.S.; Feld, G.B.; Thienel, M.; Preissl, H.; Hege, M.A.; Hallschmid, M. Oxytocin curbs calorie intake via food-specific increases in the activity of brain areas that process reward and establish cognitive control. Sci. Rep. 2018, 8, 2736. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Petersson, M.; Uvnäs-Moberg, K. Interactions of oxytocin and dopamine—Effects on behavior in health and disease. Biomedicines 2024, 12, 2440. [Google Scholar] [CrossRef]
- Swaab, D.F.; Purba, J.S.; Hofman, M.A. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: A study of five cases. J. Clin. Endocrinol. Metab. 1995, 80, 573–579. [Google Scholar]
- Swaab, D.F. Prader-Willi Syndrome and the hypothalamus. Acta Paediatr. 1997, 86 (Suppl. 423), 50–54. [Google Scholar] [CrossRef]
- Martin, A.; State, M.; Anderson, G.M.; Kaye, W.M.; Hanchett, J.M.; McConaha, C.W.; North, W.G.; Leckman, J.F. Cerebrospinal fluid levels of oxytocin in Prader–Willi syndrome: A preliminary report. Biol. Psychiatry 1998, 44, 1349–1352. [Google Scholar] [CrossRef]
- Johnson, L.; Manzardo, A.M.; Miller, J.L.; Driscoll, D.J.; Butler, M.G. Elevated plasma oxytocin levels in children with Prader–Willi syndrome compared with healthy unrelated siblings. Am. J. Med. Genet. Part A 2016, 170, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Uvnäs-Moberg, K. Role of efferent and afferent vagal nerve activity during reproduction: Integrating function of oxytocin on metabolism and behaviour. Psychoneuroendocrinology 1994, 19, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Chang, A.; Ma, R.; Strong, T.V.; Okun, M.S.; Foote, K.D.; Wexler, A.; Gunduz, A.; Miller, J.L.; Halpern, C.H. Neuromodulation for the treatment of Prader-Willi syndrome—A systematic review. Neurotherapeutics 2024, 21, e00339. [Google Scholar] [CrossRef] [PubMed]
- Meziane, H.; Schaller, F.; Bauer, S.; Villard, C.; Matarazzo, V.; Riet, F.; Guillon, G.; Lafitte, D.; Desarmenien, M.G.; Tauber, M.; et al. An early postnatal oxytocin treatment prevents social and learning deficits in adult mice deficient for Magel2, a gene involved in Prader-Willi Syndrome and autism. Biol. Psychiatry 2015, 78, 85–94. [Google Scholar] [CrossRef]
- Schaller, F.; Watrin, F.; Sturny, R.; Massacrier, A.; Szepetowski, P.; Muscatelli, F. A single postnatal injection of oxytocin rescues the lethal feeding behaviour in mouse newborns deficient for the imprinted Magel2 gene. Hum. Mol. Genet. 2010, 19, 4895–4905. [Google Scholar] [CrossRef] [PubMed]
- Petersson, M.; Höybye, C. Is Oxytocin a Contributor to Behavioral and Metabolic Features in Prader-Willi Syndrome? Curr. Issues Mol. Biol. 2024, 46, 8767–8779. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.F.; Bomberg, E.; Billington, C.; Levine, A.; Kotz, C.M. Brain-derived neurotrophic factor in the hypothalamic paraventricular nucleus reduces energy intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1003–R1012. [Google Scholar] [CrossRef]
- Queen, N.J.; Zou, X.; Anderson, J.M.; Huang, W.; Appana, B.; Komatineni, S.; Wevrick, R.; Cao, L. Hypothalamic AAV-BDNF gene therapy improves metabolic function and behavior in the Magel2-null mouse model of Prader-Willi syndrome. Mol. Ther. Methods Clin. Dev. 2022, 27, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Esteba-Castillo, S.; Novell, R.; Giménez-Palop, O.; Coronas, R.; Gabau, E.; Corripio, R.; Baena, N.; Viñas-Jornet, M.; Guitart, M.; et al. Lack of Postprandial Peak in Brain-Derived Neurotrophic Factor in Adults with Prader-Willi Syndrome. PLoS ONE 2016, 11, e0163468. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kozlov, S.V.; Bogenpohl, J.W.; Howell, M.P.; Wevrick, R.; Panda, S.; Hogenesch, J.B.; Muglia, L.J.; Van Gelder, R.N.; Herzog, E.D.; Stewart, C.L. The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 2007, 39, 1266–1272. [Google Scholar] [CrossRef]
- Manzardo, A.M.; Johnson, L.; Miller, J.L.; Driscoll, D.J.; Butler, M.G. Higher plasma orexin a levels in children with Prader-Willi syndrome compared with healthy unrelated sibling controls. Am. J. Med. Genet. A 2016, 170, 2328–2333. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gimez-Palop, O.; Casamitjana, L.; Corripio, R.; Estab-Castillo, S.; Pareja, R.; Albinana, N.; Rigla, M.; Caixas, A. Growth Hormone (GH) Treatment Decreases Plasma Kisspeptin Levels in GH-Deficient Adults with Prader-Willi Syndrome. J. Clin. Med. 2021, 10, 3054. [Google Scholar] [CrossRef]
- Omokawa, M.; Ayabe, T.; Nagai, T.; Imanishi, A.; Omokawa, A.; Nishino, S.; Sagawa, Y.; Shimizu, T.; Kanbayashi, T. Decline of CSF orexin (hypocretin) levels in Prader-Willi syndrome. Am. J. Med. Genet. A 2016, 170, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Clarke, H.; Dhillo, W.S.; Jayasena, C.N. Comprehensive Review on Kisspeptin and Its Role in Reproductive Disorders. Endocrinol. Metab. 2015, 30, 124–141. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Butler, M.G.; Nelson, T.A.; Driscoll, D.J.; Manzardo, A.M. Evaluation of plasma substance P and beta-endorphin levels in children with Prader.Willi syndrome. J. Rare Disord. 2015, 3, 1–13. [Google Scholar] [PubMed]
- Öztürk Özkan, G. Effects of Nesfatin-1 on Food Intake and Hyperglycemia. J. Am. Coll. Nutr. 2020, 39, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Gajewska, J.; Szamotulska, K.; Klemarczyk, W.; Chełchowska, M.; Strucińska, M.; Ambroszkiewicz, J. Circulating Levels of Nesfatin-1 and Spexin in Children with Prader-Willi Syndrome during Growth Hormone Treatment and Dietary Intervention. Nutrients 2023, 15, 1240. [Google Scholar] [CrossRef] [PubMed]
- Grootjen, L.N.; Kerkhof, G.F.; Juriaans, A.F.; Trueba-Timmermans, D.J.; Hokken-Koelega, A.C.S. Acute stress response of the HPA-axis in children with Prader-Willi syndrome: New insights and consequences for clinical practice. Front. Endocrinol. 2023, 14, 1146680. [Google Scholar] [CrossRef] [PubMed]
- Strelnikov, K.; Debladis, J.; Salles, J.; Valette, M.; Cortadellas, J.; Tauber, M.; Barone, P. Amygdala hyperactivation relates to eating behaviour: A potential indicator of food addiction in Prader-Willi syndrome. Brain Commun. 2023, 5, fcad138. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Krashes, M.J.; Shah, B.P.; Madara, J.C.; Olson, D.P.; Strochlic, D.E.; Garfield, A.S.; Vong, L.; Pei, H.; Watabe-Uchida, M.; Uchida, N.; et al. An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 2014, 507, 238–242. [Google Scholar] [CrossRef]
- Ibrahim, K.; Earley, B.J.; Udi, S.; Nemirovski, A.; Hadar, R.; Gammal, A.; Hirsch, H.J.; Pollak, Y.; Gross, I.; Eldar-Geva, T.; et al. Targeting the endocannabinoid/CB1 receptor system for treating obesity in Prader-Willi syndrome. Mol. Metab. 2016, 5, 1187–1199. [Google Scholar] [CrossRef]
- Yuan, M.; Li, W.; Zhu, Y.; Yu, B.; Wu, J. Asprosin: A Novel Player in Metabolic Diseases. Front. Endocrinol. 2020, 11, 64. [Google Scholar] [CrossRef]
- Faienza, F.M.; Chiarito, M.; Aureli, A.; Buganza, R.; Corica, D.; Delvecchio, M.; De Sanctis, L.; Fintini, D.; Grugni, G.; Licenziati, M.R.; et al. Lack of correlation between asprosin serum levels and hyperphagic behavior in subjects with Prader-Willi Syndrome. J. Endocrinol. Investig. 2024, 1–8. [Google Scholar] [CrossRef]
- Purtell, L.; Sze, L.; Loughnan, G.; Smith, E.; Herzog, H.; Sainsbury, A.; Steinbeck, K.; Campbell, L.V.; Viardot, A. In adults with Prader-Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-1 levels. Neuropeptides 2011, 45, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Cabral, A.; López Soto, E.J.; Epelbaum, J.; Perelló, M. Is Ghrelin Synthesized in the Central Nervous System? Int. J. Mol. Sci. 2017, 18, 638. [Google Scholar] [CrossRef]
- Ferrini, F.; Salio, C.; Lossi, L.; Merighi, A. Ghrelin in central neurons. Curr. Neuropharmacol. 2009, 7, 37–49. [Google Scholar] [CrossRef]
- Miller, J.L.; Lacroix, A.; Bird, L.M.; Shoemaker, A.H.; Haqq, A.; Deal, C.L.; Clark, K.A.; Ames, M.H.; Suico, J.G.; de la Peña, A.; et al. The Efficacy, Safety, and Pharmacology of a Ghrelin O-Acyltransferase Inhibitor for the Treatment of Prader-Willi Syndrome. J. Clin. Endocrinol. Metab. 2022, 107, e2373–e2380. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Millendo Therapeutics SAS. A Phase 2b/3 Study to Evaluate the Safety, Tolerability, and Effects of Livoletide (AZP-531), an Unacylated Ghrelin Analogue, on Food-Related Behaviors in Patients with Prader-Willi Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT03790865 (accessed on 10 February 2025).
- Mahmoud, R.; Kimonis, V.; Butler, M.G. Clinical Trials in Prader-Willi Syndrome: A Review. Int. J. Mol. Sci. 2023, 24, 2150. [Google Scholar] [CrossRef] [PubMed]
- Kishore, S.; Stamm, S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 2006, 311, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Garfield, A.S.; Davies, J.R.; Burke, L.K.; Furby, H.V.; Wilkinson, L.S.; Heisler, L.K.; Isles, A.R. Increased alternate splicing of Htr2c in a mouse model for Prader-Willi syndrome leads disruption of 5HT2C receptor mediated appetite. Mol. Brain 2016, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Kishore, P.; Boucai, L.; Zhang, K.; Li, W.; Koppaka, S.; Kehlenbrink, S.; Schiwek, A.; Esterson, Y.B.; Mehta, D.; Bursheh, S.; et al. Activation of K(ATP) channels suppresses glucose production in humans. J. Clin. Investig. 2011, 121, 4916–4920. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cowen, N.; Bhatnagar, A. The Potential Role of Activating the ATP-Sensitive Potassium Channel in the Treatment of Hyperphagic Obesity. Genes 2020, 11, 450. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kimonis, V.; Surampalli, A.; Wencel, M.; Gold, J.A.; Cowen, N.M. A randomized pilot efficacy and safety trial of diazoxide choline controlled-release in patients with Prader-Willi syndrome. PLoS ONE 2019, 14, e0221615. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miller, J.L.; Gevers, E.; Bridges, N.; Yanovski, J.A.; Salehi, P.; Obrynba, K.S.; Felner, E.I.; Bird, L.M.; Shoemaker, A.H.; Angulo, M.; et al. DESTINY PWS Investigators. Diazoxide Choline Extended-Release Tablet in People With Prader-Willi Syndrome: A Double-Blind, Placebo-Controlled Trial. J. Clin. Endocrinol. Metab. 2023, 108, 1676–1685. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miller, J.L.; Gevers, E.; Bridges, N.; Yanovski, J.A.; Salehi, P.; Obrynba, K.S.; Felner, E.I.; Bird, L.M.; Shoemaker, A.H.; Angulo, M.; et al. C601/C602 Investigators. Diazoxide choline extended-release tablet in people with Prader-Willi syndrome: Results from long-term open-label study. Obesity 2024, 32, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.L.; Qin, Y.F.; Chao, Y.Q.; Hu, C.X.; Xia, F.L.; Zou, C.C. The genotype and phenotype correlation of Prader-Willi syndrome. Rare Dis. Orphan Drugs J. 2024, 3, 33. [Google Scholar] [CrossRef]

{kind=link}
| Peptide/Protein | Difference | Type of Study | References |
|---|---|---|---|
| Oxytocin | Usually low | Autopsy material, human blood, and CSF as well as animals | [35,38,39,40,41,44] |
| Orexin | High | Human blood and anjmals | [50,51] |
| Kisspeptin | High | Human blood | [52] |
| BDNF | Low | Autopsy material, animals, and human blood | [5,48,49] |
| Tachykinins | Substance P high | Human blood | [55] |
| Nesfatin-1 | High | Human blood | [57] |
| CRH | Changed activity | Human blood, MR | [58,59] |
| TRH | Low | Human blood | [3] |
| GHRH | Low | Human blood | [3] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Höybye, C.; Petersson, M. The Role of the Arcuate Nucleus in Regulating Hunger and Satiety in Prader-Willi Syndrome. Curr. Issues Mol. Biol. 2025, 47, 192. https://doi.org/10.3390/cimb47030192
Höybye C, Petersson M. The Role of the Arcuate Nucleus in Regulating Hunger and Satiety in Prader-Willi Syndrome. Current Issues in Molecular Biology. 2025; 47(3):192. https://doi.org/10.3390/cimb47030192
Chicago/Turabian StyleHöybye, Charlotte, and Maria Petersson. 2025. "The Role of the Arcuate Nucleus in Regulating Hunger and Satiety in Prader-Willi Syndrome" Current Issues in Molecular Biology 47, no. 3: 192. https://doi.org/10.3390/cimb47030192
APA StyleHöybye, C., & Petersson, M. (2025). The Role of the Arcuate Nucleus in Regulating Hunger and Satiety in Prader-Willi Syndrome. Current Issues in Molecular Biology, 47(3), 192. https://doi.org/10.3390/cimb47030192