Abstract
Plasma lipids are essential components of biological systems, transported through interactions with proteins to maintain cellular functions. These lipids exist in various forms, such as fatty acids, glycerolipids, glycerophospholipids, sphingolipids, sterols, and prenol lipids, derived from dietary intake, adipose tissue, and biosynthesis. While the association between certain fatty acids and cardiovascular diseases has been widely recognized, polyunsaturated fatty acids (PUFAs) exhibit cardioprotective effects, reducing risks of arrhythmias and heart-related mortality. This is due to their role in the production of eicosanoids, which modulate inflammation. Chronic inflammation, particularly in obesity, is significantly influenced by fatty acids, with saturated fatty acids promoting inflammation and PUFAs mitigating it. Oxylipins, bioactive molecules derived from the oxidation of PUFAs, play crucial roles in immune regulation across various organisms, including plants, fungi, and bacteria. These molecules, such as prostaglandins, leukotrienes, and resolvins, regulate immune responses during infection and inflammation. The production of oxylipins extends beyond mammals, with fungi and bacteria synthesizing these molecules to modulate immune responses, promoting both defense and pathogenesis. This review delves into the multifaceted effects of oxylipins, exploring their impact on host and microbial interactions, with a focus on their potential for therapeutic applications in modulating infection and immune response.
1. Introduction
Plasma lipids play a critical role in the body, primarily relying on their interactions with proteins to maintain their dispersion and solubility within the bloodstream. These lipids are classified into distinct groups based on their chemical properties, with simple fatty acids and similar compounds typically binding to albumin [1]. In contrast, more complex lipids are transported by specialized plasma lipoproteins, ensuring their effective distribution and function [2]. Mammals contain nearly 600 different lipid molecules, distributed across six major lipid categories: fatty acids, glycerolipids, glycerophospholipids, sphingolipids, sterols, and prenol lipids [3,4]. The fatty acids that serve as building blocks for these complex lipids originate from a combination of dietary intake, the breakdown of adipose tissue, and endogenous biosynthesis [5].
Notably, while saturated and monounsaturated fatty acids have been linked to increased risks of arrhythmias and sudden cardiac death, polyunsaturated fatty acids (PUFAs) seem to exert a protective effect against cardiovascular disease, reducing the risk of cardiac-related mortality [6,7,8]. This protective role is largely attributed to the eicosanoids produced from PUFAs, such as prostaglandins and leukotrienes, which actively participate in mitigating inflammatory responses [9]. Chronic inflammation, a common hallmark of conditions like obesity, is significantly influenced by fatty acids, with growing evidence suggesting that free fatty acids play a central role in regulating these inflammatory processes. While saturated fatty acids exacerbate the inflammatory effects of lipopolysaccharides, certain PUFAs seem to have the opposite effect, potentially dampening inflammation through their oxidized metabolites [5].
Oxylipins—bioactive molecules derived from the oxidation of PUFAs—serve as essential signaling compounds across a wide range of organisms, from prokaryotes to eukaryotes [10,11]. These molecules include a variety of bioactive compounds, such as eicosanoids (prostaglandins, leukotrienes, and thromboxanes) and resolvins. In plants, for example, oxylipins like jasmonic acid are central to regulating growth and defense mechanisms [12], much as eicosanoids regulate immune responses in mammals. Among mammals, the oxidation of arachidonic acid (AA) is one of the most extensively studied pathways, leading to the synthesis of a wide array of eicosanoids that regulate immune function and inflammation. AA is stored in membrane phospholipids and is released at elevated levels upon cell activation, where it is metabolized into bioactive mediators like prostaglandins and leukotrienes through a series of specific enzymatic pathways [13,14]. These enzymes, known as oxygenases, are divided into three primary classes: cyclooxygenases (COXs), lipoxygenases (LOXs), and monooxygenases [15]. Together, they facilitate the conversion of PUFAs into various oxidized products (Figure 1), although alternative radical-driven pathways also contribute to oxylipin production [16]. Both the COX and LOX pathways are well-established routes in the metabolism of AA, and research has shown that the production of oxylipins is not exclusive to mammals; plants, bacteria, and fungi also produce these molecules, suggesting that they play a widespread and evolutionarily conserved role in communication and regulation, from immunity to developmental processes.
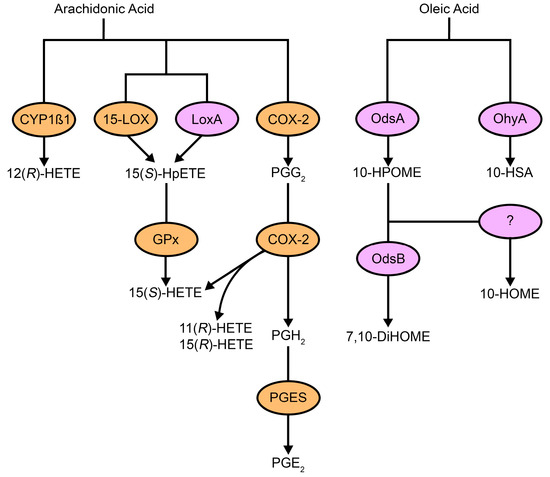
Figure 1.
Host and microbial pathways for oxylipin production. Host cells and pathogens use unsaturated fatty acid substrates, such as arachidonic acid and oleic acid, to produce oxylipins. Some oxylipins are made by both prokaryotic (purple) and eukaryotic (orange) enzymes, while others are specific to one or the other. Host-derived fatty acids also contribute to microbial pathways. Abbreviations: CYP1β1, cytochrome P450 family 1 subfamily B member 1; 15-LOX, arachidonate 15-lipoxygenase; LoxA, secretable arachidonate 15-lipoxygenase; COX-2, cyclooxygenase 2; Gpx, glutathione peroxidase; PGES, prostaglandin E synthase; OdsA, oxylipin-dependent quorum-sensing system A; OdsB, oxylipin-dependent quorum-sensing system B; OhyA, oleate hydratase; HETE, hydroxyeicosatetraenoic acid; HpETE, hydroperoxyeicosatetraenoic acid; PG, prostaglandin; HPOME, hydroperoxyoctadecenoic acid; HSA, hydroxystearic acid; DiHOME, dihydroxyoctadecenoic acid; HOME, hydroxyoctadecenoic acid.
Interestingly, the production of oxylipins is not limited to mammals. Pathogenic microbes, including bacteria and fungi, also synthesize oxylipins, either from exogenous fatty acids or through de novo pathways, to manipulate host immune responses and promote infection. This review aims to address several key gaps in the current understanding of oxylipins in microbial pathogenesis. Specifically, we will provide evidence that both pathogenic fungi and bacteria produce oxylipins and demonstrate the critical role of these oxylipins in pathogenesis. Microbial oxylipins closely resemble host-derived oxylipins, suggesting that pathogen-derived oxylipins may mimic host molecules to help pathogens regulate immune responses. By exploring these points, we aim to highlight the potential of oxylipins as novel therapeutic targets in the treatment of infectious diseases.
2. Oxylipin Immunomodulation
Oxylipins are integral mediators in the regulation of inflammatory responses during both injury and infection [17]. One of the most studied groups of oxylipins, prostaglandins, are locally produced lipids that can either promote or resolve inflammation, depending on the specific context, the type of cell involved, and the anatomical site of action. Inflammatory responses often lead to hyperalgesia, an enhanced sensitivity to pain, which serves as a hallmark symptom of inflammation [18]. Prostaglandins E2 (PGE2) [19] and I2 [20] have been particularly implicated in the potentiation of thermal hyperalgesia in experimental mouse models. These prostaglandins enhance the activation of the capsaicin receptor, a key player in the sensation of pain [21]. Macrophages, a central component of the immune system, produce these prostaglandins in response to pro-inflammatory signals, such as bacterial lipopolysaccharides. Interestingly, reverse-transcription polymerase chain reaction experiments have shown that PGE2 not only induces the production of pro-inflammatory cytokines like tumor necrosis factor alpha (TNF-α), interleukin 1 alpha (IL-1α), and IL-6 but also exerts a regulatory effect by suppressing the production of other cytokines, such as IL-1β and IL-8 [22,23]. This dual functionality underscores the complex nature of prostaglandins in mediating inflammation.
PGE2 plays complex and often contradictory roles in immune responses, influencing various conditions such as cancer, autoimmune diseases, and infections. In cancer, PGE2 suppresses antitumor immunity by inhibiting the expansion and differentiation of CD8+ T cells within the tumor microenvironment through EP2/EP4 signaling [24,25]. This inhibition promotes immune evasion and tumor progression. However, blocking PGE2 signaling can restore T-cell function and enhance anticancer immunity [25], suggesting that targeting PGE2 may offer therapeutic benefits. In autoimmune diseases, PGE2 contributes to the expansion and activation of Th17 cells, which play a role in conditions such as multiple sclerosis and rheumatoid arthritis [26,27]. Despite this, PGE2 also has a regulatory role, suppressing Th1 responses and promoting the resolution of inflammation, which may help restore tissue homeostasis and prevent further damage in autoimmune disorders [26,28]. During infections, PGE2 can dampen the host’s immune response, as seen in diseases like tuberculosis, by suppressing innate immunity and facilitating pathogen survival [29,30]. On the other hand, PGE2 can also play a protective role by promoting anti-inflammatory macrophage phenotypes, helping to limit excessive inflammation and preventing tissue damage during infection [30,31].
The mechanistic basis for how PGE2 exhibits both pro-inflammatory and anti-inflammatory effects depends on its interaction with four receptor subtypes (EP1-EP4). These receptors can mediate opposite effects, with the concentration of PGE2 and the type of cell involved determining the outcome. For instance, PGE2 promotes inflammation through several mechanisms. It enhances vascular permeability and promotes leukocyte infiltration, both of which contribute to inflammation [32,33]. It also activates mast cells through the EP3 receptor [33], worsening the inflammatory response. Additionally, PGE2 can inhibit regulatory T cells (Tregs) through EP4, activating the immune system to respond to the microbiota and fostering intestinal inflammation [34]. At low concentrations, PGE2 stimulates pro-inflammatory cytokine production by activation of nuclear factor-kappa B (NF-κB) [31].
However, PGE2 also has anti-inflammatory effects. It can suppress Type 2 inflammation by inhibiting eosinophil chemotaxis and limiting Th2 cell activation [35,36]. At higher concentrations, PGE2 inhibits NF-κB activity, reducing pro-inflammatory cytokine production and promoting the resolution of inflammation [31]. Furthermore, PGE2 induces the development of FOXP3+ regulatory T cells through the EP4 receptor, contributing to immune suppression and tissue homeostasis.
The impact of PGE2 extends beyond pain modulation; it also significantly influences immune responses. For instance, exposure to PGE2 has been shown to promote the migration of Langerhans cells—specialized dendritic cells found in the skin—to regional lymph nodes. Although this migration enhances the T-cell stimulatory capacity of Langerhans cells, it also simultaneously suppresses CD4+ T-cell proliferation [37], highlighting the nuanced effects of PGE2 on the immune system. Building on this reasoning, parasite-derived prostaglandin D2 has been observed to activate epidermal Langerhans cells in the context of skin infections [38]. This activation causes their retention within the epidermis, impairing their ability to migrate to other sites. As a result, the skin’s contact hypersensitivity response is diminished, which helps limit excessive inflammation.
Interestingly, in a guinea pig model of contact dermatitis, PGE2 is produced on the skin upon exposure to allergens [39], further linking prostaglandins to skin immune responses. Additionally, a human keratinocyte skin cell line produces leukotriene B4, as well as 12- and 15-HETE, when exposed to bacterial Actinobacilli species in vitro [40]. Supernatant from this skin cell line after bacterial exposure significantly enhances the production of PGE2 and other oxylipins by neutrophils compared to neutrophils stimulated with supernatants from unexposed skin cells [40]. These findings underscore the role of oxylipins in modulating immune responses during bacterial skin infections and inflammation.
Beyond prostaglandins, other oxylipins such as hydroxy fatty acids, including 9- and 13-HODE (hydroxyoctadecadienoic acid), are byproducts of linoleic acid peroxidation and are found to accumulate in inflammatory disease states. In patients with progressive rheumatoid arthritis, for instance, 9-HODE levels in low-density lipoprotein particles increase significantly [41]. Northern blotting assays have revealed that 9-HODE induces IL-1β mRNA expression in human macrophages [42]. Additionally, enzyme-linked immunosorbent assays confirm that both 9-HODE and 13-HODE can stimulate the release of IL-1β cytokines from these cells, further emphasizing the impact of these bioactive lipids on the inflammatory cascade.
This section underscores the complexity and versatility of oxylipins, particularly prostaglandins and hydroxylated fatty acids, in modulating immune responses. Their ability to both promote and resolve inflammation, depending on the context, highlights their importance as central players in the regulation of immune function. The ongoing research into these molecules continues to reveal their diverse functions in various pathological conditions, offering potential therapeutic avenues for managing chronic inflammatory diseases.
3. Fungal Oxylipins
Most fungi do not naturally produce significant quantities of AA in the way that mammals do, so they acquire environmental AA and convert it into various oxidized products through enzymatic pathways [43,44]. Additionally, fungi synthesize oxylipins common to both plants and animals, such as 9(S)-HODE and 13(S)-HODE, which are produced by the LOX pathway [45]. This ability to synthesize oxylipins is shared between fungi and mammals, with both groups utilizing eicosanoids—such as prostaglandins and leukotrienes—as key mediators in immune modulation and other physiological processes [45,46,47,48] (Figure 1 and Figure 2).
Fungal pathogens, particularly those involved in human infections, often manipulate oxylipin production to their advantage, using these molecules to regulate host immune responses and promote infection. For example, Candida albicans, a fungus normally present in the human microbiota, can become pathogenic under certain conditions, leading to life-threatening infections [49]. The immune system generally restricts the overgrowth of C. albicans in the intestines, but the fungus can circumvent these defenses by producing eicosanoids like PGE2. In the presence of human keratinocytes, C. albicans and C. tropicalis produce significantly higher levels of PGE2, which suppresses the expression of interferon γ-inducible protein 10 (IP-10), a molecule involved in the inflammatory response [50]. This suppression of IP-10 may impair the body’s ability to mount an effective response to a cutaneous fungal infection.

Figure 2.
Oxylipins in microbial pathogenesis and host signaling. Host immune cells produce oxylipins like PGE2, which promote immunosuppressive effects, and 15-HETE, which regulates immune responses. The lipid mediators are actively secreted during infection or inflammation, balancing pro- and anti-inflammatory factors [28,29,51,52,53]. Pathogens (e.g., Pseudomonas aeruginosa, Staphylococcus aureus, and Candida albicans) produce and use oxylipins to manipulate host immune responses, mimicking host regulation. PGE2 (orange) is produced by fungi and host cells, while 15-HETE (purple) is made by bacteria and host cells. Abbreviations: LPS, liposaccharide; IL-1β, interleukin 1 beta; TNF-α, tumor necrosis factor alpha; PG, prostaglandin; HETE, hydroxyeicosatetraenoic acid; HSA, hydroxystearic acid.
Figure 2.
Oxylipins in microbial pathogenesis and host signaling. Host immune cells produce oxylipins like PGE2, which promote immunosuppressive effects, and 15-HETE, which regulates immune responses. The lipid mediators are actively secreted during infection or inflammation, balancing pro- and anti-inflammatory factors [28,29,51,52,53]. Pathogens (e.g., Pseudomonas aeruginosa, Staphylococcus aureus, and Candida albicans) produce and use oxylipins to manipulate host immune responses, mimicking host regulation. PGE2 (orange) is produced by fungi and host cells, while 15-HETE (purple) is made by bacteria and host cells. Abbreviations: LPS, liposaccharide; IL-1β, interleukin 1 beta; TNF-α, tumor necrosis factor alpha; PG, prostaglandin; HETE, hydroxyeicosatetraenoic acid; HSA, hydroxystearic acid.

Prostaglandins, particularly PGE2, play a critical role in modulating the immune response during infection. By suppressing Th1-type immune responses while promoting a Th2-type response [54,55,56,57], PGE2 helps establish a favorable environment for the pathogen, thereby limiting the ability of macrophages to clear C. albicans and other fungal species [58,59]. Interestingly, although C. albicans lacks a COX homolog, nonspecific synthetic oxygenase inhibitors, but not COX-specific synthetic inhibitors, can block PGE2 production in the fungus [60]. This suggests that C. albicans may utilize alternative enzymatic pathways to produce prostaglandins, possibly through enzymes like stearyl-coenzyme A desaturase (Ole2) and multicopper ferroxidase (Fet3) [60]. Ole2 does not directly affect AA or prostaglandins; however, it likely alters upstream fatty acids, supplying substrates or intermediates used for PGE2 synthesis [60,61]. Fet3 may regulate iron homeostasis, affecting enzymes involved in PGE2 synthesis [61], and mutants deficient in Fet3 have reduced PGE2 levels [60,61]. The deletion of these enzymes does not completely abolish PGE2 production [60,61], indicating additional pathways exist in fungal prostaglandin biosynthesis. Importantly, the absence of Ole2 impairs C. albicans’ ability to colonize the gastrointestinal tract and makes the fungus more susceptible to macrophage phagocytosis [61], underscoring the impact of oxylipins on virulence [62]. Ole2 and Fet3 are key components of a unique fungal pathway for prostaglandin synthesis, though their roles appear to be indirect or regulatory rather than catalytic. Further research is needed to clarify this pathway. Additionally, PGE2 signaling suppresses CD4+ T-cell activation in the lamina propria and the development of colitis in a mouse model of dextran sodium sulfate-induced colitis [63]. This anti-inflammatory mechanism may help explain, at least in part, how Candida species can colonize the gut, as it dampens the immune response, allowing the fungus to thrive in the gastrointestinal environment.
In addition to prostaglandins, C. albicans also synthesizes resolvin E1 (RvE1), an anti-inflammatory oxylipin that reduces neutrophil migration in response to IL-8 [64]. RvE1 plays a complex role in fungal infections, and its biosynthesis is blocked by LOX and monooxygenase inhibitors. In a mouse model of systemic candidiasis, RvE1 enhances neutrophil phagocytosis and promotes the production of reactive oxygen species [64], which are critical for fungal clearance. However, the impact of RvE1 on fungal virulence appears to be dose-dependent, with low concentrations of RvE1 conferring protection, and high concentrations potentially driving an excessive immune response.
The regulation of RvE1 involves selective receptor interactions, immune cell modulation, and context-dependent signaling. RvE1 primarily acts through two receptors: ChemR23 and BLT1. As an agonist for ChemR23 on macrophages and dendritic cells, RvE1 promotes anti-inflammatory and pro-resolving effects by reducing cytokine production and enhancing phagocytosis [65,66]. In contrast, RvE1 antagonizes BLT1, a receptor for leukotriene B4 (LTB4), to suppress neutrophil recruitment and actin polymerization, key processes in inflammation [65,66]. RvE1 also modulates cytokine levels, inhibiting pro-inflammatory cytokines like IL-6, IL-17, and IL-23, while boosting anti-inflammatory cytokines such as TGF-β. In dendritic cells and Th17 cells, RvE1 prevents Th17 differentiation and reduces their migration by downregulating CCR6 expression [65,67]. Additionally, RvE1 limits dendritic cell motility by inhibiting LTB4-BLT1 signaling [65], essential for actin polymerization and migration, thus reducing dendritic cell-driven T-cell activation in immune responses. RvE1’s role in inflammation resolution is evident in its ability to enhance microbial clearance and prevent excessive tissue damage. For instance, it reduces neutrophil infiltration and inflammatory mediators like IL-1β and MCP-1 in acute lung injury models [68]. Through these mechanisms, RvE1 balances immune responses, resolving inflammation while maintaining tissue homeostasis. Its effects are context-dependent, shaped by receptor expression, cellular targets, and inflammatory signals.
Pathogenic yeasts such as C. neoformans also produce oxylipins, including leukotrienes and prostaglandins, even in the absence of exogenous AA supplementation. C. neoformans produces a dehydrogenated form of PGE2, known as 15-keto-PGE2, which activates the host nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ), promoting intracellular survival during macrophage infection [69]. Inhibition of 15-keto-PGE2 production impairs fungal survival, emphasizing the importance of oxylipin-mediated immune modulation in fungal pathogenesis.
In addition to these effects, fungal prostaglandins have been implicated in regulating the yeast-to-hyphal transition in C. albicans and C. neoformans [70]. This transition is crucial for fungal virulence, as it allows the fungi to disseminate throughout the body, invade tissues, and form biofilms [71,72]. In the yeast form, these fungi can adhere to endothelial cells and spread throughout the bloodstream [73,74,75]. PGE2 produced during hyphal formation modulates host immunity by reducing pro-inflammatory cytokines like TNF-α and increasing anti-inflammatory cytokines like IL-10, aiding fungal evasion and persistence [70,72]. However, in the hyphal form, they become more adept at evading the host immune system and infiltrating tissues [76]. COX inhibitors like aspirin block hyphal formation and biofilm development in C. albicans [72,77], and mutants that are unable to undergo this transition exhibit significantly reduced infectivity [78,79,80], further supporting the critical effect of oxylipin-mediated signaling in fungal virulence. Thus, the fungus synthesizes PGE2 from AA, typically sourced from host lipids, to utilize host resources for regulating its morphogenesis and survival [61,70].
In Aspergillus species, such as Aspergillus nidulans and A. fumigatus, oxylipins also play a key role in fungal pathogenesis. These species encode three COX orthologs (ppoABC) that contribute to prostaglandin biosynthesis [81]. In a mouse model of invasive pulmonary aspergillosis, a triple knockout mutant lacking these COX orthologs demonstrated increased resistance to oxidative stress and hypervirulence. Interestingly, prostaglandins in these fungi have different effects, with PGE2 inhibiting A. nidulans asexual sporulation (important for fungal dissemination) and PGE2 also inhibiting pigment formation in A. fumigatus hyphae. Although spore pigmentation and gliotoxin production are known to be key virulence factors in A. fumigatus [82,83,84], neither of these traits is affected in the ppo knockout mutant. This suggests that prostaglandin biosynthesis may contribute to virulence through an additional, yet unidentified, mechanism. These observations highlight the complexity of oxylipin signaling in fungal pathogens, suggesting that prostaglandins may modulate fungal growth and virulence in species-specific ways.
4. Bacterial Oxylipins
Bacteria, much like plants, animals, and fungi, produce oxylipins and oxylipin-like molecules that serve as essential signaling molecules, modulating a variety of physiological processes within both the bacteria themselves and their host organisms (Figure 1 and Figure 2). These molecules are critical in regulating bacterial growth, host interactions, and immune responses, often influencing the progression of infections. One well-studied example is Pseudomonas aeruginosa, a Gram-negative opportunistic pathogen commonly involved in mixed infections with C albicans [85]. It is a major contributor to morbidity and mortality in individuals with cystic fibrosis or those who are immunocompromised [86,87,88].
P. aeruginosa is capable of synthesizing oxylipins through the incorporation of host-derived AA. Using its LOX ortholog, LoxA, P. aeruginosa produces 15(R)-hydroxyeicosatetraenoic acid (15(R)-HETE), a key oxylipin that influences immune modulation [89,90,91]. Interestingly, 15(R)-HETE plays an important role in inducing anti-inflammatory effects in the host by promoting the synthesis of lipoxins, which are known to facilitate the resolution of inflammation [89,92]. Neutrophils, once they internalize 15(R)-HETE, convert this molecule into the R configuration of lipoxins (e.g., 15-epi-LXA4), which actively resolve inflammation and aid in the restoration of tissue homeostasis [93]. In contrast, the S stereoisomer of 15-HETE, which is naturally found in mammals, is converted into a less potent form of lipoxin (e.g., LXA4) that has a shorter half-life and a diminished anti-inflammatory effect [94,95]. There is evidence that P. aeruginosa is capable of synthesizing prostaglandins [96], but more work is needed to determine their precise impact on pathogenesis. COX-like enzymes, identified in bacterial systems, play a role in PGE2 production. While specific enzymes analogous to mammalian cyclooxygenases have not been fully characterized in P. aeruginosa, studies suggest that bacterial cyclooxygenase-like enzymes contribute to PGE2 synthesis [97]. This ability to modulate inflammation through the synthesis of oxylipins demonstrates how bacteria have evolved to manipulate host immune responses in ways that facilitate their survival and pathogenesis.
The presence of lipoxygenases in bacteria was first identified with the discovery of the loxA gene in P. aeruginosa. Since then, lipoxygenases have been characterized in other bacterial species including Nitrosomonas europaea and Anabaena sp. strain PCC 7120, neither of which are known to be infectious [89]. Nonpathogenic bacteria use oxylipins as autoinducers in quorum sensing to regulate gene expression based on cell density [98]. This system, distinct from traditional quorum sensing mechanisms, involves LysR-type transcriptional regulators. Oxylipins also influence bacterial motility, biofilm formation, and multicellular behavior, enhancing survival under stress [99]. Additionally, they enable bacteria to interact with plants and fungi, impacting host defense mechanisms or symbiotic relationships. For instance, bacterial oxylipins can mimic plant signaling molecules to modulate plant immunity [100]. By regulating transitions between planktonic and biofilm states, oxylipins help bacteria adapt to harsh environments [100,101], underscoring their role in bacterial communication, even in nonpathogenic species. In mammals, lipoxins like 15-HETE have important functions as signaling molecules in immune regulation. Observations demonstrate that P. aeruginosa secretes these oxylipins into the periplasm and extracellular spaces; however, further research is needed to fully understand the implications of this process and its impact on bacterial pathogenesis.
P. aeruginosa also produces other oxylipins, such as 10(S)-hydroxy-(8E)-octadecenoic acid (10(S)-HOME) and 7S,10S-dihydroxy-(8E)-octadecenoic acid (7,10-DiHOME) [98,99]. These molecules are synthesized by 10-S Dioxygenase (OdsA) and 7,10-Diol Synthase (OdsB), which are encoded by the PA2077 and PA2078 genes, respectively [102]. Both genes are part of an operon that also includes PA2076, which encodes OdsR, a probable transcriptional regulator. The discovery of this oxylipin biosynthetic pathway highlights a novel quorum-sensing system, known as the ODS (oxylipin-dependent quorum sensing system), that regulates bacterial motility and biofilm formation [98]. Production of 10(S)-HOME and 7,10-DiHOME involves secretion of OdsAB through the Xcp Type II secretion system [103], which is a distinctive feature compared to other quorum-sensing systems. These processes are essential for P. aeruginosa to establish persistent infections, particularly in cystic fibrosis patients where biofilms play a significant role in chronic lung infections [99,104]. Quorum sensing is a mechanism by which bacteria communicate with each other to coordinate group behaviors, such as the formation of biofilms, which increase bacterial resistance to host immune responses and antimicrobial treatments [105]. This process is driven by oxylipins derived from host oleic acid, which are essential for effective biofilm formation and pathogenicity [103]. By producing these oxylipins, P. aeruginosa can regulate its own gene expression, promoting biofilm formation and improving survival in hostile environments [98,99].
In vitro studies revealed that these oxylipins promote microcolony formation both over abiotic surfaces and biotic surfaces, including monolayers of A549 human alveolar epithelial cells, which P. aeruginosa infects in cystic fibrosis patients [106]. Furthermore, in vivo testing in Drosophila melanogaster showed that a P. aeruginosa strain with knockout mutations of both OdsA and OdsB had half the mortality rate of the wild-type strain when inoculated using the pricking method [99]. Further investigations revealed that P. aeruginosa senses the presence of 10(S)-HOME and 7,10-DiHOME based on cell density using the ODS system. These oxylipins induce the expression of their own biosynthetic operon [98]. When extracellular levels of oxylipins reach a certain threshold, they bind to OdsR, which upregulates the production of more OdsA and OdsB enzymes. These enzymes then convert additional PUFAs into oxylipins that are subsequently detected by an unknown receptor within the cell [98]. This feedback loop ensures that the quorum-sensing system is activated in response to rising oxylipin concentrations. Notably, the system requires the presence of exogenous oleic acid to activate OdsR, ensuring that the ODS system is triggered only in environments containing this fatty acid, which is typically found in living organisms. This suggests that P. aeruginosa has evolved to utilize host-derived oleic acid as a strategy for establishing successful infections [107]. The discovery of oxylipin production and its significance in bacterial quorum sensing, along with the identification of homologs for OdsA and OdsB across various Pseudomonas species and other bacterial genera, suggests that targeting oxylipin-mediated communication could offer new therapeutic opportunities to disrupt bacterial quorum sensing and biofilm formation, potentially providing novel treatments for chronic infections.
Beyond P. aeruginosa, other bacterial pathogens also utilize oxylipins in their interactions with hosts. Staphylococcus aureus, a Gram-positive bacterium commonly associated with skin infections and a frequent co-colonizer with C. albicans in cystic fibrosis patients [108,109], also exploits oxylipins to modulate immune responses. S. aureus can metabolize host-derived oleic acid into 10(R)-hydroxystearic acid (10-HSA) [110], a hydroxylated fatty acid that plays a crucial role in modulating immune responses during infection. Increased levels of oleic acid are correlated with more severe cystic fibrosis disease progression [111], and S. aureus exploits this by producing 10-HSA. This molecule activates PPARα, which suppresses the host’s innate immune response, facilitating immune evasion and enhancing the bacterium’s survival [112].
In addition, 10-HSA is abundant in the pulmonary discharge from cystic fibrosis patients infected with P. aeruginosa or S. aureus [113], providing further evidence of its impact on infection. P. aeruginosa may produce 10-HSA [114] through at least two distinct pathways: the LoxA enzyme and the OdsAB system. In contrast, S. aureus relies solely on the enzyme oleate hydratase (OhyA) to convert oleic acid from the membrane bilayer into 10-HSA [115,116,117,118]. OhyA is involved in a larger metabolic program to metabolize environmental fatty acids [119], but genetic disruption of ohyA does not significantly affect S. aureus growth in vitro [110]. Nevertheless, S. aureus’s ability to produce 10-HSA remains crucial for its virulence, as evidenced by the impaired infection capability in a mouse model when ohyA is disrupted [120]. These findings underscore the importance of oxylipin production in bacterial pathogenesis.
Furthermore, Streptococcus pyogenes, a bacterium responsible for flesh-eating disease, also encodes the OhyA enzyme and produces hydroxylated fatty acids, such as 10-HSA. In S. pyogenes, the production of these fatty acids enhances bacterial virulence by promoting adherence to host tissues and facilitating internalization into host cells. A knockout strain of S. pyogenes that cannot produce these hydroxylated fatty acids exhibits reduced virulence, as it is less capable of adhering to and internalizing into host keratinocytes [121]. These findings suggest that bacterial production of oxylipins not only helps modulate the immune system but also enhances pathogen fitness by facilitating critical processes such as adherence, internalization, and evasion of immune detection. Interestingly, many bacterial taxa, including probiotic beneficial symbionts from the gut microbiome, also encode the enzyme OhyA, which plays a role in oxylipin metabolism [122,123,124,125]. This suggests that OhyA may contribute to host tolerance of the gut microbiome and support symbiosis.
Another notable bacterium, Mycobacterium tuberculosis, a highly pathogenic bacterium responsible for tuberculosis, also manipulates the balance of oxylipins to escape immune detection and modulate the host’s inflammatory response. One of the key oxylipins involved in this process is PGE2, which is induced by M. tuberculosis during infection. PGE2 plays a dual role in modulating the immune response, as it suppresses pro-inflammatory cytokine production while simultaneously promoting autophagy in infected cells [29,126]. This autophagic response helps to contain the bacteria within the host cell, thereby limiting the spread of infection. However, M. tuberculosis also induces the synthesis of lipoxin A4 (LXA4), which triggers necrosis of infected cells, enabling the bacteria to escape and disseminate throughout the host [127,128]. This complex interplay between PGE2 and LXA4 influences the outcome of the infection, with excessive LXA4 promoting bacterial survival and dissemination through necrotic cell death [129,130]. Thus, the balance of oxylipins plays a critical role in determining the success of M. tuberculosis infection and the progression of tuberculosis.
5. Targeting Oxylipins in Therapy
The role of oxylipins in microbial colonization and infection remains unclear. In particular, it is uncertain how many distinct microbes rely on oxylipins for these processes. Another unresolved question in polymicrobial settings is whether all members of the microbial community need to produce oxylipins, or if production by just a few members can protect the entire community.
Several therapeutic strategies aim to target oxylipins by modulating their production, signaling pathways, or downstream effects in order to treat infections and inflammatory diseases. These strategies range from broad-spectrum approaches to highly targeted ones. For example, oxylipin biosynthesis can be inhibited using COX and LOX inhibitors. Nonsteroidal anti-inflammatory drugs (e.g., aspirin) inhibit COX enzymes, reducing prostaglandin production in both the host and pathogens like C. albicans [131,132]. LOX inhibitors can block bacterial exploitation of host-derived oxylipins, as seen in P. aeruginosa and the plant pathogen Xylella fastidiosa [100,133]. Another approach involves blocking oxylipin signaling through EP receptor antagonists. For example, blocking receptors like EP4 has been shown to reduce prostaglandin-mediated immunosuppression in cancer and infections [131,132], thus enhancing immune responses. Additionally, synthetic oxylipin analogs can be used to disrupt signaling. Modified oxylipins inspired by natural products have been developed to inhibit biofilm formation in pathogens like S. aureus by disrupting quorum sensing and biofilm matrix production [134].
A major concern in the use of broad-spectrum antimicrobial treatments is the collateral damage to the microbiome. Recent advancements in narrow-spectrum therapeutics have shown promise in creating pathogen-specific therapies [135]. One example is inhibiting P. aeruginosa diol synthase enzymes to block the synthesis of virulence-enhancing oxylipins like 10-HOME and 7,10-DiHOME [99,136]. Another approach that may help preserve the microbiome involves targeting oxylipins through diet. Supplementation with omega-3 fatty acids alters the oxylipin profile, favoring anti-inflammatory mediators such as resolvins, which counteract pro-inflammatory bacterial lipids [131,132]. Modifying gut microbiota through diet or probiotics can also influence oxylipin production and reduce inflammation-related diseases [132].
However, translating oxylipin-targeting therapies into clinical practice faces several challenges. Analytical and technical difficulties related to standardization, lipid complexity, and rapid turnover pose significant obstacles. Oxylipins are metabolically unstable with short half-lives, making it challenging to accurately measure their levels and effects in vivo [137]. Furthermore, variability in sample collection, preparation, and analysis across laboratories complicates the generation of reproducible data. To overcome these challenges, harmonized protocols and reference materials are essential for establishing consistent oxylipin profiles [137,138].
The complexity of oxylipin signaling pathways, which involve overlapping and redundant functions, makes it difficult to isolate their specific roles. Biological variability also adds complexity, as oxylipins can have pro-inflammatory or anti-inflammatory effects depending on tissue type, disease state, or concentration [138,139]. This variability makes therapeutic targeting difficult. Genetic, metabolic, and environmental factors also influence oxylipin production and response [138], highlighting the need for personalized approaches to treatment. Many oxylipins interact with multiple receptors or pathways, which increases the risk of unintended side effects, such as impaired platelet function or excessive immune suppression [137,140]. Moreover, developing stable formulations that effectively deliver oxylipin-modulating agents to target tissues without causing systemic side effects remains a challenge [137].
Despite these hurdles, targeting oxylipins holds significant potential for treating inflammatory diseases, infections, and cardiovascular conditions. Although few clinical trials have tested oxylipin-targeted therapies, soluble epoxide hydrolase inhibitors for cardiovascular diseases show promise and warrant further validation in diverse populations [141]. To overcome the challenges in oxylipin-based therapies, standardized protocols for oxylipin profiling must be developed. Additionally, large-scale cohort studies are needed to validate biomarkers and therapeutic targets, and advances in drug delivery systems and bioinformatics tools are necessary to enhance the precision of oxylipin-based treatments.
6. Conclusions
Oxylipins, bioactive lipids derived from polyunsaturated fatty acids, play critical roles in modulating immune responses, inflammation, and microbial pathogenesis in both fungi and bacteria. These molecules are involved in host–pathogen interactions, influencing microbial virulence, biofilm formation, immune evasion, and host immune regulation. In fungi, oxylipins like prostaglandins and resolvins help pathogens such as C. albicans and A. fumigatus manipulate immune responses and promote infection persistence. Similarly, bacteria such as P. aeruginosa and S. aureus exploit oxylipins to enhance pathogenicity, while beneficial gut microbiota also contribute to the oxylipin pool.
The therapeutic potential of targeting oxylipins is significant, with strategies ranging from inhibiting biosynthesis pathways (e.g., COX and LOX inhibitors) to modulating signaling through receptor antagonists and synthetic analogs. However, challenges remain in translating these strategies into clinical practice due to issues such as metabolic instability, biological variability, and potential side effects.
Future research should focus on overcoming these challenges by developing standardized protocols for oxylipin profiling, improving drug delivery systems and creating more precise narrow-spectrum treatments to avoid disrupting the microbiome. Additionally, investigating oxylipin metabolism in the gut microbiome could offer new avenues for managing intestinal dysbiosis and enhancing microbiome health. Further studies into the concentration-dependent effects of oxylipins and their interactions with multiple microbial species will be crucial in refining therapeutic approaches and identifying reliable biomarkers for infection diagnosis and treatment outcomes.
Author Contributions
R.J.N. and C.D.R. conceptualization and investigation; R.J.N. writing—original draft; R.J.N. and C.D.R. writing—reviewing and editing; C.D.R. resources, project administration, and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Institutes of Health Grant R00-AI166116 (C.D.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- van der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Havel, R.J.; Eder, H.A.; Bragdon, J.H. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Investig. 1955, 34, 1345–1353. [Google Scholar] [CrossRef]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef]
- Quehenberger, O.; Dennis, E.A. The human plasma lipidome. N. Engl. J. Med. 2011, 365, 1812–1823. [Google Scholar] [CrossRef]
- Leaf, A. Plasma nonesterified fatty acid concentration as a risk factor for sudden cardiac death: The Paris Prospective Study. Circulation 2001, 104, 744–745. [Google Scholar] [CrossRef] [PubMed]
- McLennan, P.L. Relative effects of dietary saturated, monounsaturated, and polyunsaturated fatty acids on cardiac arrhythmias in rats. Am. J. Clin. Nutr. 1993, 57, 207–212. [Google Scholar] [CrossRef]
- Burr, M.L.; Fehily, A.M.; Gilbert, J.F.; Rogers, S.; Holliday, R.M.; Sweetnam, P.M.; Elwood, P.C.; Deadman, N.M. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: Diet and reinfarction trial (DART). Lancet 1989, 2, 757–761. [Google Scholar] [CrossRef]
- Buczynski, M.W.; Dumlao, D.S.; Dennis, E.A. Thematic review series: Proteomics. An integrated omics analysis of eicosanoid biology. J. Lipid Res. 2009, 50, 1015–1038. [Google Scholar] [CrossRef]
- Du, Y.; Taylor, C.G.; Aukema, H.M.; Zahradka, P. Role of oxylipins generated from dietary PUFAs in the modulation of endothelial cell function. Prostaglandins Leukot. Essent. Fatty Acids 2020, 160, 102160. [Google Scholar] [CrossRef]
- Andreou, A.; Brodhun, F.; Feussner, I. Biosynthesis of oxylipins in non-mammals. Prog. Lipid Res. 2009, 48, 148–170. [Google Scholar] [CrossRef] [PubMed]
- Carvalhais, L.C.; Dennis, P.G.; Badri, D.V.; Tyson, G.W.; Vivanco, J.M.; Schenk, P.M. Activation of the jasmonic acid plant defence pathway alters the composition of rhizosphere bacterial communities. PLoS ONE 2013, 8, e56457. [Google Scholar] [CrossRef]
- Konya, V.; Mjosberg, J. Lipid mediators as regulators of human ILC2 function in allergic diseases. Immunol. Lett. 2016, 179, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Beermann, C.; Neumann, S.; Fussbroich, D.; Zielen, S.; Schubert, R. Combinations of distinct long-chain polyunsaturated fatty acid species for improved dietary treatment against allergic bronchial asthma. Nutrition 2016, 32, 1165–1170. [Google Scholar] [CrossRef]
- Noverr, M.C.; Erb-Downward, J.R.; Huffnagle, G.B. Production of eicosanoids and other oxylipins by pathogenic eukaryotic microbes. Clin. Microbiol. Rev. 2003, 16, 517–533. [Google Scholar] [CrossRef]
- Durand, T.; Bultel-Ponce, V.; Guy, A.; Berger, S.; Mueller, M.J.; Galano, J.M. New bioactive oxylipins formed by non-enzymatic free-radical-catalyzed pathways: The phytoprostanes. Lipids 2009, 44, 875–888. [Google Scholar] [CrossRef]
- Samuchiwal, S.K.; Boyce, J.A. Role of lipid mediators and control of lymphocyte responses in type 2 immunopathology. J. Allergy Clin. Immunol. 2018, 141, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef]
- Reddy, S.T.; Herschman, H.R. Ligand-induced prostaglandin synthesis requires expression of the TIS10/PGS-2 prostaglandin synthase gene in murine fibroblasts and macrophages. J. Biol. Chem. 1994, 269, 15473–15480. [Google Scholar] [CrossRef]
- Tang, Y.; Di Pietro, L.; Feng, Y.; Wang, X. Increased TNF-a and PGI2, but not NO release from macrophages in 18-month-old rats. Mech. Ageing Dev. 2000, 114, 79–88. [Google Scholar] [CrossRef]
- Moriyama, T.; Higashi, T.; Togashi, K.; Iida, T.; Segi, E.; Sugimoto, Y.; Tominaga, T.; Narumiya, S.; Tominaga, M. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol. Pain 2005, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.W.; Burke, P.A.; Drotar, M.E.; Chavali, S.R.; Forse, R.A. Effects of prostaglandin E2, cholera toxin and 8-bromo-cyclic AMP on lipopolysaccharide-induced gene expression of cytokines in human macrophages. Immunology 1995, 84, 446–452. [Google Scholar] [PubMed]
- Ikegami, R.; Sugimoto, Y.; Segi, E.; Katsuyama, M.; Karahashi, H.; Amano, F.; Maruyama, T.; Yamane, H.; Tsuchiya, S.; Ichikawa, A. The expression of prostaglandin E receptors EP2 and EP4 and their different regulation by lipopolysaccharide in C3H/HeN peritoneal macrophages. J. Immunol. 2001, 166, 4689–4696. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and cancer: Insight into tumor progression and immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Lacher, S.B.; Dorr, J.; de Almeida, G.P.; Honninger, J.; Bayerl, F.; Hirschberger, A.; Pedde, A.M.; Meiser, P.; Ramsauer, L.; Rudolph, T.J.; et al. PGE2 limits effector expansion of tumour-infiltrating stem-like CD8(+) T cells. Nature 2024, 629, 417–425. [Google Scholar] [CrossRef]
- Burkett, J.B.; Doran, A.C.; Gannon, M. Harnessing prostaglandin E2 signaling to ameliorate autoimmunity. Trends Immunol. 2023, 44, 162–171. [Google Scholar] [CrossRef]
- Kofler, D.M.; Marson, A.; Dominguez-Villar, M.; Xiao, S.; Kuchroo, V.K.; Hafler, D.A. Decreased RORC-dependent silencing of prostaglandin receptor EP2 induces autoimmune Th17 cells. J. Clin. Investig. 2014, 124, 2513–2522. [Google Scholar] [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef]
- Pellegrini, J.M.; Martin, C.; Morelli, M.P.; Schander, J.A.; Tateosian, N.L.; Amiano, N.O.; Rolandelli, A.; Palmero, D.J.; Levi, A.; Ciallella, L.; et al. PGE2 displays immunosuppressive effects during human active tuberculosis. Sci. Rep. 2021, 11, 13559. [Google Scholar] [CrossRef]
- Agard, M.; Asakrah, S.; Morici, L.A. PGE2 suppression of innate immunity during mucosal bacterial infection. Front. Cell. Infect. Microbiol. 2013, 3, 45. [Google Scholar] [CrossRef]
- Martin-Vazquez, E.; Cobo-Vuilleumier, N.; Lopez-Noriega, L.; Lorenzo, P.I.; Gauthier, B.R. The PTGS2/COX2-PGE2 signaling cascade in inflammation: Pro or anti? A case study with type 1 diabetes mellitus. Int. J. Biol. Sci. 2023, 19, 4157–4165. [Google Scholar] [CrossRef] [PubMed]
- Gomez, I.; Foudi, N.; Longrois, D.; Norel, X. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot. Essent. Fatty Acids 2013, 89, 55–63. [Google Scholar] [CrossRef]
- Kawahara, K.; Hohjoh, H.; Inazumi, T.; Tsuchiya, S.; Sugimoto, Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta 2015, 1851, 414–421. [Google Scholar] [CrossRef]
- Crittenden, S.; Goepp, M.; Pollock, J.; Robb, C.T.; Smyth, D.J.; Zhou, Y.; Andrews, R.; Tyrrell, V.; Gkikas, K.; Adima, A.; et al. Prostaglandin E2 promotes intestinal inflammation via inhibiting microbiota-dependent regulatory T cells. Sci. Adv. 2021, 7, eabd7954. [Google Scholar] [CrossRef]
- Oyesola, O.O.; Tait Wojno, E.D. Prostaglandin regulation of type 2 inflammation: From basic biology to therapeutic interventions. Eur. J. Immunol. 2021, 51, 2399–2416. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Maher, S.A.; Dekkak, B.; Jones, V.; Wong, S.; Brook, P.; Belvisi, M.G. Anti-inflammatory effects of PGE2 in the lung: Role of the EP4 receptor subtype. Thorax 2015, 70, 740–747. [Google Scholar] [CrossRef]
- Kabashima, K.; Sakata, D.; Nagamachi, M.; Miyachi, Y.; Inaba, K.; Narumiya, S. Prostaglandin E2-EP4 signaling initiates skin immune responses by promoting migration and maturation of Langerhans cells. Nat. Med. 2003, 9, 744–749. [Google Scholar] [CrossRef]
- Angeli, V.; Faveeuw, C.; Roye, O.; Fontaine, J.; Teissier, E.; Capron, A.; Wolowczuk, I.; Capron, M.; Trottein, F. Role of the parasite-derived prostaglandin D2 in the inhibition of epidermal Langerhans cell migration during schistosomiasis infection. J. Exp. Med. 2001, 193, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, T.; Printz, M.P. Arachidonic acid metabolism in skin: Experimental contact dermatitis in guinea pigs. Int. Arch. Allergy Appl. Immunol. 1982, 69, 347–352. [Google Scholar] [CrossRef]
- Eberhard, J.; Jepsen, S.; Pohl, L.; Albers, H.K.; Acil, Y. Bacterial challenge stimulates formation of arachidonic acid metabolites by human keratinocytes and neutrophils in vitro. Clin. Diagn. Lab. Immunol. 2002, 9, 132–137. [Google Scholar] [CrossRef]
- Jira, W.; Spiteller, G.; Richter, A. Increased levels of lipid oxidation products in low density lipoproteins of patients suffering from rheumatoid arthritis. Chem. Phys. Lipids 1997, 87, 81–89. [Google Scholar] [CrossRef]
- Ku, G.; Thomas, C.E.; Akeson, A.L.; Jackson, R.L. Induction of interleukin 1 beta expression from human peripheral blood monocyte-derived macrophages by 9-hydroxyoctadecadienoic acid. J. Biol. Chem. 1992, 267, 14183–14188. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.D.; Klug, M.J. Characterization and differentiation of filamentous fungi based on fatty acid composition. Appl. Environ. Microbiol. 1996, 62, 4136–4146. [Google Scholar] [CrossRef]
- Ells, R.; Kemp, G.; Albertyn, J.; Kock, J.L.; Pohl, C.H. Phenothiazine is a potent inhibitor of prostaglandin E2 production by Candida albicans biofilms. FEMS Yeast Res. 2013, 13, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.J.; Keller, N.P. Production of cross-kingdom oxylipins by pathogenic fungi: An update on their role in development and pathogenicity. J. Microbiol. 2016, 54, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, A.I.; Sa-Nunes, A.; Soares, E.G.; Peres, C.M.; Silva, C.L.; Faccioli, L.H. Blockade of endogenous leukotrienes exacerbates pulmonary histoplasmosis. Infect. Immun. 2004, 72, 1637–1644. [Google Scholar] [CrossRef]
- Secatto, A.; Rodrigues, L.C.; Serezani, C.H.; Ramos, S.G.; Dias-Baruffi, M.; Faccioli, L.H.; Medeiros, A.I. 5-Lipoxygenase deficiency impairs innate and adaptive immune responses during fungal infection. PLoS ONE 2012, 7, e31701. [Google Scholar] [CrossRef]
- Nicolete, R.; Secatto, A.; Pereira, P.A.; Soares, E.G.; Faccioli, L.H. Leukotriene B4-loaded microspheres as a new approach to enhance antimicrobial responses in Histoplasma capsulatum-infected mice. Int. J. Antimicrob. Agents 2009, 34, 365–369. [Google Scholar] [CrossRef]
- Odds, F.C. Candida infections: An overview. Crit. Rev. Microbiol. 1987, 15, 1–5. [Google Scholar] [CrossRef]
- Shiraki, Y.; Ishibashi, Y.; Hiruma, M.; Nishikawa, A.; Ikeda, S. Candida albicans abrogates the expression of interferon-g-inducible protein-10 in human keratinocytes. FEMS Immunol. Med. Microbiol. 2008, 54, 122–128. [Google Scholar] [CrossRef]
- Sheppe, A.E.F.; Edelmann, M.J. Roles of eicosanoids in regulating inflammation and neutrophil migration as an innate host response to bacterial infections. Infect. Immun. 2021, 89, e0009521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sun, K.; Wang, W.S. Identification of a feed-forward loop between 15(S)-HETE and PGE2 in human amnion at parturition. J. Lipid Res. 2022, 63, 100294. [Google Scholar] [CrossRef]
- Salari, H.; Chan-Yeung, M. Release of 15-hydroxyeicosatetraenoic acid (15-HETE) and prostaglandin E2 (PGE2) by cultured human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1989, 1, 245–250. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Betz, M.; Fox, B.S. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 1991, 146, 108–113. [Google Scholar] [CrossRef]
- Romani, L.; Kaufmann, S.H. Immunity to fungi: Editorial overview. Res. Immunol. 1998, 149, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Noverr, M.C.; Toews, G.B.; Huffnagle, G.B. Production of prostaglandins and leukotrienes by pathogenic fungi. Infect. Immun. 2002, 70, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.P.; van de Veerdonk, F.L.; van der Meer, J.W.; Kullberg, B.J.; Joosten, L.A.; Netea, M.G. The Candida Th17 response is dependent on mannan- and b-glucan-induced prostaglandin E2. Int. Immunol. 2010, 22, 889–895. [Google Scholar] [CrossRef]
- Shen, L.; Liu, Y. Prostaglandin E2 blockade enhances the pulmonary anti-Cryptococcus neoformans immune reaction via the induction of TLR-4. Int. Immunopharmacol. 2015, 28, 376–381. [Google Scholar] [CrossRef]
- Erb-Downward, J.R.; Noverr, M.C. Characterization of prostaglandin E2 production by Candida albicans. Infect. Immun. 2007, 75, 3498–3505. [Google Scholar] [CrossRef]
- Tan, T.G.; Lim, Y.S.; Tan, A.; Leong, R.; Pavelka, N. Fungal symbionts produce prostaglandin E2 to promote their intestinal colonization. Front. Cell. Infect. Microbiol. 2019, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, T.; Thuer, E.; Heijink, M.; Toth, R.; Bodai, L.; Vagvolgyi, C.; Giera, M.; Gabaldon, T.; Gacser, A. Eicosanoid biosynthesis influences the virulence of Candida parapsilosis. Virulence 2018, 9, 1019–1035. [Google Scholar] [CrossRef]
- Kabashima, K.; Saji, T.; Murata, T.; Nagamachi, M.; Matsuoka, T.; Segi, E.; Tsuboi, K.; Sugimoto, Y.; Kobayashi, T.; Miyachi, Y.; et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J. Clin. Investig. 2002, 109, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Haas-Stapleton, E.J.; Lu, Y.; Hong, S.; Arita, M.; Favoreto, S.; Nigam, S.; Serhan, C.N.; Agabian, N. Candida albicans modulates host defense by biosynthesizing the pro-resolving mediator resolvin E1. PLoS ONE 2007, 2, e1316. [Google Scholar] [CrossRef] [PubMed]
- Sawada, Y.; Honda, T.; Hanakawa, S.; Nakamizo, S.; Murata, T.; Ueharaguchi-Tanada, Y.; Ono, S.; Amano, W.; Nakajima, S.; Egawa, G.; et al. Resolvin E1 inhibits dendritic cell migration in the skin and attenuates contact hypersensitivity responses. J. Exp. Med. 2015, 212, 1921–1930. [Google Scholar] [CrossRef]
- Ishida, T.; Yoshida, M.; Arita, M.; Nishitani, Y.; Nishiumi, S.; Masuda, A.; Mizuno, S.; Takagawa, T.; Morita, Y.; Kutsumi, H.; et al. Resolvin E1, an endogenous lipid mediator derived from eicosapentaenoic acid, prevents dextran sulfate sodium-induced colitis. Inflamm. Bowel Dis. 2010, 16, 87–95. [Google Scholar] [CrossRef]
- Oner, F.; Alvarez, C.; Yaghmoor, W.; Stephens, D.; Hasturk, H.; Firatli, E.; Kantarci, A. Resolvin E1 regulates Th17 function and T cell activation. Front. Immunol. 2021, 12, 637983. [Google Scholar] [CrossRef]
- Seki, H.; Fukunaga, K.; Arita, M.; Arai, H.; Nakanishi, H.; Taguchi, R.; Miyasho, T.; Takamiya, R.; Asano, K.; Ishizaka, A.; et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J. Immunol. 2010, 184, 836–843. [Google Scholar] [CrossRef]
- Evans, R.J.; Pline, K.; Loynes, C.A.; Needs, S.; Aldrovandi, M.; Tiefenbach, J.; Bielska, E.; Rubino, R.E.; Nicol, C.J.; May, R.C.; et al. 15-keto-prostaglandin E2 activates host peroxisome proliferator-activated receptor gamma (PPAR-g) to promote Cryptococcus neoformans growth during infection. PLoS Pathog. 2019, 15, e1007597. [Google Scholar] [CrossRef]
- Noverr, M.C.; Phare, S.M.; Toews, G.B.; Coffey, M.J.; Huffnagle, G.B. Pathogenic yeasts Cryptococcus neoformans and Candida albicans produce immunomodulatory prostaglandins. Infect. Immun. 2001, 69, 2957–2963. [Google Scholar] [CrossRef]
- Schimanski, J.; Gresnigt, M.S.; Brunner, E.; Werz, O.; Hube, B.; Garscha, U. Hyphal-associated protein expression is crucial for Candida albicans-induced eicosanoid biosynthesis in immune cells. Eur. J. Immunol. 2024, 54, e2350743. [Google Scholar] [CrossRef] [PubMed]
- Mochochoko, B.M.; Pohl, C.H.; O’Neill, H.G. Candida albicans-enteric viral interactions-the prostaglandin E2 connection and host immune responses. iScience 2023, 26, 105870. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.M.; Gianetti, B.A.; Witchley, J.N. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat. Rev. Microbiol. 2017, 15, 96–108. [Google Scholar] [CrossRef]
- Fradin, C.; De Groot, P.; MacCallum, D.; Schaller, M.; Klis, F.; Odds, F.C.; Hube, B. Granulocytes govern the transcriptional response, morphology and proliferation of Candida albicans in human blood. Mol. Microbiol. 2005, 56, 397–415. [Google Scholar] [CrossRef]
- Grubb, S.E.; Murdoch, C.; Sudbery, P.E.; Saville, S.P.; Lopez-Ribot, J.L.; Thornhill, M.H. Adhesion of Candida albicans to endothelial cells under physiological conditions of flow. Infect. Immun. 2009, 77, 3872–3878. [Google Scholar] [CrossRef] [PubMed]
- Erwig, L.P.; Gow, N.A. Interactions of fungal pathogens with phagocytes. Nat. Rev. Microbiol. 2016, 14, 163–176. [Google Scholar] [CrossRef]
- Alem, M.A.; Douglas, L.J. Effects of aspirin and other nonsteroidal anti-inflammatory drugs on biofilms and planktonic cells of Candida albicans. Antimicrob. Agents Chemother. 2004, 48, 41–47. [Google Scholar] [CrossRef]
- Nemecek, J.C.; Wuthrich, M.; Klein, B.S. Global control of dimorphism and virulence in fungi. Science 2006, 312, 583–588. [Google Scholar] [CrossRef]
- Brown, A.J.; Gow, N.A. Regulatory networks controlling Candida albicans morphogenesis. Trends Microbiol. 1999, 7, 333–338. [Google Scholar] [CrossRef]
- Lo, H.J.; Kohler, J.R.; DiDomenico, B.; Loebenberg, D.; Cacciapuoti, A.; Fink, G.R. Nonfilamentous C. albicans mutants are avirulent. Cell 1997, 90, 939–949. [Google Scholar] [CrossRef]
- Tsitsigiannis, D.I.; Bok, J.W.; Andes, D.; Nielsen, K.F.; Frisvad, J.C.; Keller, N.P. Aspergillus cyclooxygenase-like enzymes are associated with prostaglandin production and virulence. Infect. Immun. 2005, 73, 4548–4559. [Google Scholar] [CrossRef]
- Brakhage, A.A.; Langfelder, K.; Wanner, G.; Schmidt, A.; Jahn, B. Pigment biosynthesis and virulence. Contrib. Microbiol. 1999, 2, 205–215. [Google Scholar] [CrossRef]
- Tsai, H.F.; Chang, Y.C.; Washburn, R.G.; Wheeler, M.H.; Kwon-Chung, K.J. The developmentally regulated alb1 gene of Aspergillus fumigatus: Its role in modulation of conidial morphology and virulence. J. Bacteriol. 1998, 180, 3031–3038. [Google Scholar] [CrossRef]
- Nieminen, S.M.; Maki-Paakkanen, J.; Hirvonen, M.R.; Roponen, M.; von Wright, A. Genotoxicity of gliotoxin, a secondary metabolite of Aspergillus fumigatus, in a battery of short-term test systems. Mutat. Res. 2002, 520, 161–170. [Google Scholar] [CrossRef]
- Kaleli, I.; Cevahir, N.; Demir, M.; Yildirim, U.; Sahin, R. Anticandidal activity of Pseudomonas aeruginosa strains isolated from clinical specimens. Mycoses 2007, 50, 74–78. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Hayes, D., Jr.; Wozniak, D.J. Cystic fibrosis and Pseudomonas aeruginosa: The host-microbe interface. Clin. Microbiol. Rev. 2019, 32, e00138-00118. [Google Scholar] [CrossRef] [PubMed]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Vance, R.E.; Hong, S.; Gronert, K.; Serhan, C.N.; Mekalanos, J.J. The opportunistic pathogen Pseudomonas aeruginosa carries a secretable arachidonate 15-lipoxygenase. Proc. Natl. Acad. Sci. USA 2004, 101, 2135–2139. [Google Scholar] [CrossRef]
- Banthiya, S.; Kalms, J.; Galemou Yoga, E.; Ivanov, I.; Carpena, X.; Hamberg, M.; Kuhn, H.; Scheerer, P. Structural and functional basis of phospholipid oxygenase activity of bacterial lipoxygenase from Pseudomonas aeruginosa. Biochim. Biophys. Acta 2016, 1861, 1681–1692. [Google Scholar] [CrossRef]
- Deschamps, J.D.; Ogunsola, A.F.; Jameson, J.B., 2nd; Yasgar, A.; Flitter, B.A.; Freedman, C.J.; Melvin, J.A.; Nguyen, J.V.; Maloney, D.J.; Jadhav, A.; et al. Biochemical and cellular characterization and inhibitor discovery of Pseudomonas aeruginosa 15-lipoxygenase. Biochemistry 2016, 55, 3329–3340. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxin biosynthesis: An update and role in anti-inflammation and pro-resolution. Prostaglandins Other Lipid Mediat. 2002, 68–69, 433–455. [Google Scholar] [CrossRef]
- Serhan, C.N. Lipoxins and novel aspirin-triggered 15-epi-lipoxins (ATL): A jungle of cell-cell interactions or a therapeutic opportunity? Prostaglandins 1997, 53, 107–137. [Google Scholar] [CrossRef]
- Gewirtz, A.T.; McCormick, B.; Neish, A.S.; Petasis, N.A.; Gronert, K.; Serhan, C.N.; Madara, J.L. Pathogen-induced chemokine secretion from model intestinal epithelium is inhibited by lipoxin A4 analogs. J. Clin. Investig. 1998, 101, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Fiore, S.; Maddox, J.F.; Brady, H.R.; Petasis, N.A.; Serhan, C.N. Aspirin-triggered 15-epi-lipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: Evidence for anti-inflammatory receptors. J. Exp. Med. 1997, 185, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Lamacka, M.; Sajbidor, J. The occurrence of prostaglandins and related compounds in lower organisms. Prostaglandins Leukot. Essent. Fatty Acids 1995, 52, 357–364. [Google Scholar] [CrossRef]
- Turinsky, J.; Loegering, D.J. Prostaglandin E2 and muscle protein turnover in Pseudomonas aeruginosa sepsis. Biochim. Biophys. Acta 1985, 840, 137–140. [Google Scholar] [CrossRef]
- Martinez, E.; Cosnahan, R.K.; Wu, M.; Gadila, S.K.; Quick, E.B.; Mobley, J.A.; Campos-Gomez, J. Oxylipins mediate cell-to-cell communication in Pseudomonas aeruginosa. Commun. Biol. 2019, 2, 66. [Google Scholar] [CrossRef]
- Martinez, E.; Campos-Gomez, J. Oxylipins produced by Pseudomonas aeruginosa promote biofilm formation and virulence. Nat. Commun. 2016, 7, 13823. [Google Scholar] [CrossRef]
- Beccaccioli, M.; Pucci, N.; Salustri, M.; Scortichini, M.; Zaccaria, M.; Momeni, B.; Loreti, S.; Reverberi, M.; Scala, V. Fungal and bacterial oxylipins are signals for intra- and inter-cellular communication within plant disease. Front. Plant Sci. 2022, 13, 823233. [Google Scholar] [CrossRef]
- Kurakin, G.F. Bacterial oxylipins: A key to multicellularity and to combating antimicrobial resistance? Priroda 2022, 26–32. [Google Scholar] [CrossRef]
- Estupinan, M.; Alvarez-Garcia, D.; Barril, X.; Diaz, P.; Manresa, A. In silico/in vivo insights into the functional and evolutionary pathway of Pseudomonas aeruginosa oleate-diol synthase. discovery of a new bacterial di-heme cytochrome c peroxidase subfamily. PLoS ONE 2015, 10, e0131462. [Google Scholar] [CrossRef]
- Martinez, E.; Orihuela, C.J.; Campos-Gomez, J. Pseudomonas aeruginosa secretes the oxylipin autoinducer synthases OdsA and OdsB via the Xcp type 2 secretion system. J. Bacteriol. 2022, 204, e0011422. [Google Scholar] [CrossRef]
- Burrows, L.L. Pseudomonas aeruginosa twitching motility: Type IV pili in action. Annu. Rev. Microbiol. 2012, 66, 493–520. [Google Scholar] [CrossRef]
- Pohl, C.H.; Kock, J.L. Oxidized fatty acids as inter-kingdom signaling molecules. Molecules 2014, 19, 1273–1285. [Google Scholar] [CrossRef]
- Ciofu, O.; Hansen, C.R.; Hoiby, N. Respiratory bacterial infections in cystic fibrosis. Curr. Opin. Pulm. Med. 2013, 19, 251–258. [Google Scholar] [CrossRef]
- Oliveira, A.F.; Cunha, D.A.; Ladriere, L.; Igoillo-Esteve, M.; Bugliani, M.; Marchetti, P.; Cnop, M. In vitro use of free fatty acids bound to albumin: A comparison of protocols. Biotechniques 2015, 58, 228–233. [Google Scholar] [CrossRef]
- Valenza, G.; Tappe, D.; Turnwald, D.; Frosch, M.; Konig, C.; Hebestreit, H.; Abele-Horn, M. Prevalence and antimicrobial susceptibility of microorganisms isolated from sputa of patients with cystic fibrosis. J. Cyst. Fibros. 2008, 7, 123–127. [Google Scholar] [CrossRef]
- Bauernfeind, A.; Bertele, R.M.; Harms, K.; Horl, G.; Jungwirth, R.; Petermuller, C.; Przyklenk, B.; Weisslein-Pfister, C. Qualitative and quantitative microbiological analysis of sputa of 102 patients with cystic fibrosis. Infection 1987, 15, 270–277. [Google Scholar] [CrossRef]
- Subramanian, C.; Frank, M.W.; Batte, J.L.; Whaley, S.G.; Rock, C.O. Oleate hydratase from Staphylococcus aureus protects against palmitoleic acid, the major antimicrobial fatty acid produced by mammalian skin. J. Biol. Chem. 2019, 294, 9285–9294. [Google Scholar] [CrossRef]
- Campbell, I.M.; Crozier, D.N.; Caton, R.B. Abnormal fatty acid composition and impaired oxygen supply in cystic fibrosis patients. Pediatrics 1976, 57, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Radka, C.D.; Frank, M.W.; Simmons, T.S.; Johnson, C.N.; Rosch, J.W.; Rock, C.O. Staphylococcus aureus oleate hydratase produces ligands that activate host PPARa. Front. Cell. Infect. Microbiol. 2024, 14, 1352810. [Google Scholar] [CrossRef]
- Campbell, I.M.; Crozier, D.N.; Trim, A.; Sigrist, J. Cystic fibrosis and bacterial conversion of oleic acid to a cathartic, 10-hydroxystearic acid. Lancet 1987, 2, 107. [Google Scholar] [CrossRef]
- Wallen, L.L.; Benedict, R.G.; Jackson, R.W. The microbiological production of 10-hydroxystearic acid from oleic acid. Arch. Biochem. Biophys. 1962, 99, 249–253. [Google Scholar] [CrossRef]
- Radka, C.D.; Grace, C.R.; Hasdemir, H.S.; Li, Y.; Rodriguez, C.C.; Rodrigues, P.; Oldham, M.L.; Qayyum, M.Z.; Pitre, A.; MacCain, W.J.; et al. The carboxy terminus causes interfacial assembly of oleate hydratase on a membrane bilayer. J. Biol. Chem. 2024, 300, 105627. [Google Scholar] [CrossRef]
- Oldham, M.L.; Zuhaib Qayyum, M.; Kalathur, R.C.; Rock, C.O.; Radka, C.D. Cryo-EM reconstruction of oleate hydratase bound to a phospholipid membrane bilayer. J. Struct. Biol. 2024, 216, 108116. [Google Scholar] [CrossRef]
- Lathram, W.A.; Neff, R.J.; Zalla, A.N.; Brien, J.D.; Subramanian, V.; Radka, C.D. Dissecting the biophysical mechanisms of oleate hydratase association with membranes. Front. Mol. Biosci. 2024, 11, 1504373. [Google Scholar] [CrossRef]
- Radka, C.D.; Batte, J.L.; Frank, M.W.; Young, B.M.; Rock, C.O. Structure and mechanism of Staphylococcus aureus oleate hydratase (OhyA). J. Biol. Chem. 2021, 296, 100252. [Google Scholar] [CrossRef]
- Radka, C.D. Interfacial enzymes enable Gram-positive microbes to eat fatty acids. Membranes 2023, 13, 423. [Google Scholar] [CrossRef]
- Radka, C.D.; Batte, J.L.; Frank, M.W.; Rosch, J.W.; Rock, C.O. Oleate hydratase (OhyA) is a virulence determinant in Staphylococcus aureus. Microbiol. Spectr. 2021, 9, e0154621. [Google Scholar] [CrossRef]
- Volkov, A.; Liavonchanka, A.; Kamneva, O.; Fiedler, T.; Goebel, C.; Kreikemeyer, B.; Feussner, I. Myosin cross-reactive antigen of Streptococcus pyogenes M49 encodes a fatty acid double bond hydratase that plays a role in oleic acid detoxification and bacterial virulence. J. Biol. Chem. 2010, 285, 10353–10361. [Google Scholar] [CrossRef]
- Neff, R.J.; Lages, P.C.; Donworth, S.K.; Brien, J.D.; Radka, C.D. Independent evolution of oleate hydratase clades in Bacillales reflects molecular convergence. Front. Mol. Biosci. 2024, 11, 1485485. [Google Scholar] [CrossRef]
- Volkov, A.; Khoshnevis, S.; Neumann, P.; Herrfurth, C.; Wohlwend, D.; Ficner, R.; Feussner, I. Crystal structure analysis of a fatty acid double-bond hydratase from Lactobacillus acidophilus. Acta Crystallogr. D. Biol. Crystallogr. 2013, 69, 648–657. [Google Scholar] [CrossRef]
- Rosberg-Cody, E.; Liavonchanka, A.; Gobel, C.; Ross, R.P.; O’Sullivan, O.; Fitzgerald, G.F.; Feussner, I.; Stanton, C. Myosin-cross-reactive antigen (MCRA) protein from Bifidobacterium breve is a FAD-dependent fatty acid hydratase which has a function in stress protection. BMC Biochem. 2011, 12, 9. [Google Scholar] [CrossRef]
- Zhao, G.; Kempen, P.J.; Shetty, R.; Gu, L.; Zhao, S.; Ruhdal Jensen, P.; Solem, C. Harnessing cross-resistance-sustainable nisin production from low-value food side streams using a Lactococcus lactis mutant with higher nisin-resistance obtained after prolonged chlorhexidine exposure. Bioresour. Technol. 2022, 348, 126776. [Google Scholar] [CrossRef]
- Park, H.S.; Choi, S.; Back, Y.W.; Lee, K.I.; Choi, H.G.; Kim, H.J. Mycobacterium tuberculosis RpfE-induced prostaglandin E2 in dendritic cells induces Th1/Th17 cell differentiation. Int. J. Mol. Sci. 2021, 22, 7535. [Google Scholar] [CrossRef]
- Pavan Kumar, N.; Moideen, K.; Nancy, A.; Viswanathan, V.; Shruthi, B.S.; Shanmugam, S.; Hissar, S.; Kornfeld, H.; Babu, S. Plasma eicosanoid levels in tuberculosis and tuberculosis-diabetes co-morbidity are associated with lung pathology and bacterial burden. Front. Cell. Infect. Microbiol. 2019, 9, 335. [Google Scholar] [CrossRef]
- Nore, K.G.; Jorgensen, M.J.; Dyrhol-Riise, A.M.; Jenum, S.; Tonby, K. Elevated levels of anti-inflammatory eicosanoids and monocyte heterogeneity in Mycobacterium tuberculosis infection and disease. Front. Immunol. 2020, 11, 579849. [Google Scholar] [CrossRef]
- Kaushal, D. Eicosanoids, prostaglandins, and the progression of tuberculosis. J. Infect. Dis. 2012, 206, 1803–1805. [Google Scholar] [CrossRef]
- Sorgi, C.A.; Soares, E.M.; Rosada, R.S.; Bitencourt, C.S.; Zoccal, K.F.; Pereira, P.A.T.; Fontanari, C.; Brandao, I.; Masson, A.P.; Ramos, S.G.; et al. Eicosanoid pathway on host resistance and inflammation during Mycobacterium tuberculosis infection is comprised by LTB4 reduction but not PGE2 increment. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165574. [Google Scholar] [CrossRef]
- Gretschel, J.; El Hage, R.; Wang, R.; Chen, Y.; Pietzner, A.; Loew, A.; Leineweber, C.G.; Wordemann, J.; Rohwer, N.; Weylandt, K.H.; et al. Harnessing oxylipins and inflammation modulation for prevention and treatment of colorectal cancer. Int. J. Mol. Sci. 2024, 25, 5408. [Google Scholar] [CrossRef]
- Misheva, M.; Johnson, J.; McCullagh, J. Role of oxylipins in the inflammatory-related diseases NAFLD, obesity, and type 2 diabetes. Metabolites 2022, 12, 1238. [Google Scholar] [CrossRef]
- Savchenko, T.; Degtyaryov, E.; Radzyukevich, Y.; Buryak, V. Therapeutic potential of plant oxylipins. Int. J. Mol. Sci. 2022, 23, 14627. [Google Scholar] [CrossRef]
- Peran, J.E.; Salvador-Reyes, L.A. Modified oxylipins as inhibitors of biofilm formation in Staphylococcus epidermidis. Front. Pharmacol. 2024, 15, 1379643. [Google Scholar] [CrossRef]
- Radka, C.D.; Rock, C.O. Mining fatty acid biosynthesis for new antimicrobials. Annu. Rev. Microbiol. 2022, 76, 281–304. [Google Scholar] [CrossRef]
- Niu, M.; Keller, N.P. Co-opting oxylipin signals in microbial disease. Cell. Microbiol. 2019, 21, e13025. [Google Scholar] [CrossRef]
- Alotaibi, A. Oxylipins in health and disease: Overcoming analytical, temporal, and functional obstacles. J. Clin. Res. Bioeth. 2024, 15, 506. [Google Scholar] [CrossRef]
- Gladine, C.; Fedorova, M. The clinical translation of eicosanoids and other oxylipins, although challenging, should be actively pursued. J. Mass Spectrom. Adv. Clin. Lab 2021, 21, 27–30. [Google Scholar] [CrossRef]
- Biagini, D.; Franzini, M.; Oliveri, P.; Lomonaco, T.; Ghimenti, S.; Bonini, A.; Vivaldi, F.; Macera, L.; Balas, L.; Durand, T.; et al. MS-based targeted profiling of oxylipins in COVID-19: A new insight into inflammation regulation. Free Radic. Biol. Med. 2022, 180, 236–243. [Google Scholar] [CrossRef]
- Stanger, L.; Yamaguchi, A.; Yalavarthi, P.; Lambert, S.; Gilmore, D.; Rickenberg, A.; Luke, C.; Kumar, K.; Obi, A.T.; White, A.; et al. The oxylipin analog CS585 prevents platelet activation and thrombosis through activation of the prostacyclin receptor. Blood 2023, 142, 1556–1569. [Google Scholar] [CrossRef]
- Hateley, C.; Olona, A.; Halliday, L.; Edin, M.L.; Ko, J.H.; Forlano, R.; Terra, X.; Lih, F.B.; Beltran-Debon, R.; Manousou, P.; et al. Multi-tissue profiling of oxylipins reveal a conserved up-regulation of epoxide:diol ratio that associates with white adipose tissue inflammation and liver steatosis in obesity. eBioMedicine 2024, 103, 105127. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).