
A Rare Case of Upper Gastrointestinal Bleeding: Osler-Weber-Rendu Syndrome



Abstract
:1. Introduction
2. HHT Diagnosis
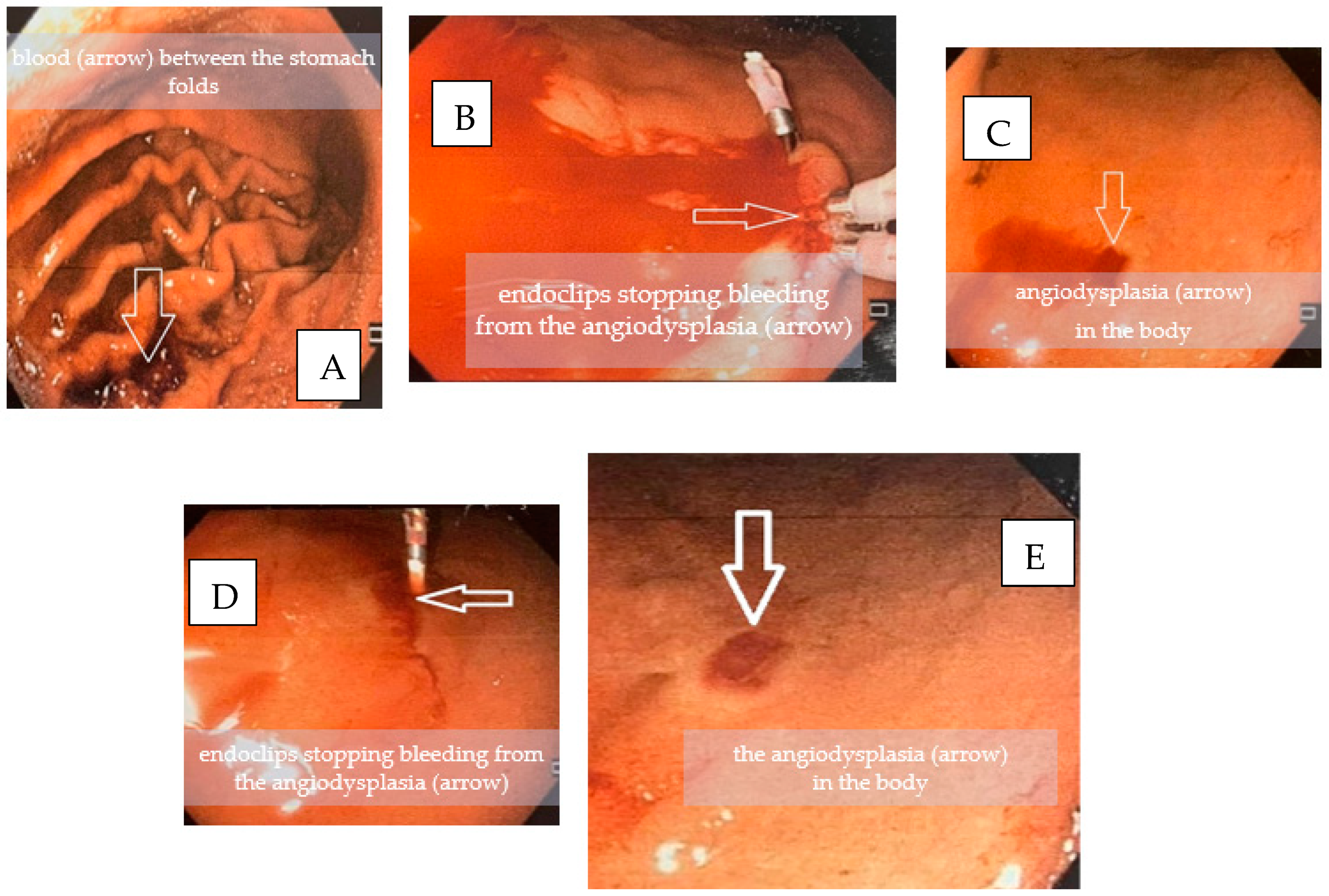
3. Case Report
4. Discussion
4.1. Epistaxis
4.2. Visceral/Gastrointestinal Telangiectasias
4.3. Hepatic AVMs
4.4. Pulmonary AVMs
4.5. Cerebral AVMs and Spinal AVMs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tortora, A.; Riccioni, M.E.; Gaetani, E.; Ojetti, V.; Holleran, G.; Gasbarrini, A. Rendu-Osler-Weber disease: A gastroenterologist’s perspective. Orphanet J. Rare Dis. 2019, 14, 130. [Google Scholar] [CrossRef] [Green Version]
- Shovlin, C.L. Pulmonary arteriovenous malformations. Am. J. Respir. Crit. Care Med. 2014, 190, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Alicea-Guevara, R.; Cruz Caliz, M.; Adorno, J.; Fernandez, R.; Rivera, K.; Gonzalez, G.; Hernandez-Castillo, R.A.; Fernandez, R.; Latorre, C.C. Life-threatening hemoptysis: Case of Osler-Weber-Rendu Syndrome. Oxf. Med. Case Rep. 2018, 22, omx108. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5865522/ (accessed on 6 June 2021). [CrossRef]
- Kritharis, A.; Al-Samkari, H.; Kuter, D.J. Hereditary hemorrhagic telangiectasia: Diagnosis and management from the hematologist’s perspective. Haematologica 2018, 103, 1433–1443. [Google Scholar] [CrossRef]
- Jackson, S.B.; Villano, N.P.; Benhammou, J.N.; Lewis, M.; Pisegna, J.R.; Padua, D. Gastrointestinal Manifestations of Hereditary Hemorrhagic Telangiectasia (HHT): A Systematic Review of the Literature. Dig. Dis. Sci. 2017, 62, 2623–2630. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, Y.; Iqbal, I.Z.; Puvanendran, M.; ElBadawey, M.R.; Carrie, S. A postal survey of hereditary hemorrhagic telangectasia in the northeast of England. Allergy Rhinol. 2015, 6, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Eker, O.F.; Boccardi, E.; Sure, U.; Patel, M.C.; Alicante, S.; Alsafi, A.; Coote, N.; Droege, F.; Dupuis, O.; Fialla, A.D.; et al. European Reference Network for Rare Vascular Diseases (VASCERN) position statement on cerebral screening in adults and children with hereditary haemorrhagic telangiectasia (HHT). Orphanet J. Rare Dis. 2020, 15, 165. [Google Scholar] [CrossRef]
- Kilian, A.; Latino, G.A.; White, A.J.; Clark, D.; Chakinala, M.M.; Ratjen, F.; McDonald, J.; Whitehead, K.J.; Gossage, J.R.; Lin, D.; et al. Genotype–Phenotype Correlations in Children with HHT. J. Clin. Med. 2020, 9, 2714. [Google Scholar] [CrossRef]
- Gaetani, E.; Agostini, F.; Giarretta, I.; Porfidia, A.; Di Martino, L.; Gasbarrini, A.; Pola, R. Antithrombotic Therapy in Hereditary Hemorrhagic Telangiectasia: Real-World Data from the Gemelli Hospital HHT Registry. J. Clin. Med. 2020, 9, 1699. [Google Scholar] [CrossRef]
- Bernabeu, C.; Bayrak-Toydemir, P.; McDonald, J.; Letarte, M. Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 3571. [Google Scholar] [CrossRef]
- Van Tuyl, S.A.C.; Letteboer, T.G.W.; Rogge-Wolf, C.; Kuipers, E.J.; Snijder, R.J.; Westermann, C.J.J.; Stolk, M.F.J. Assessment of intestinal vascular malformations in patients with hereditary hemorrhagic teleangiectasia and anemia. Eur. J. Gastroenterol. Hepatol. 2007, 19, 153–158. [Google Scholar] [CrossRef]
- Canzonieri, C.; Centenara, L.; Ornati, F.; Pagella, F.; Matti, E.; Alvisi, C.; Danesino, C.; Perego, M.; Olivieri, C. Endoscopic evaluation of gastrointestinal tract in patients with hereditary hemorrhagic telangiectasia and correlation with their genotypes. Genet. Med. 2014, 16, 3–10. [Google Scholar] [CrossRef]
- Lesca, G.; Olivieri, C.; Burnichon, N.; Pagella, F.; Carette, M.-F.; Gilbert-Dussardier, B.; Goizet, C.; Roume, J.; Rabilloud, M.; Saurin, J.-C.; et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: Data from the French-Italian HHT network. Genet. Med. 2007, 9, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Sabbà, C.; Pasculli, G.; Lenato, G.M.; Suppressa, P.; Lastella, P.; Memeo, M.; Dicuonzo, F.; Guanti, G. Hereditary hemorrhagic telangiectasia: Clinical features in ENG and ALK1 mutation carriers. J. Thromb. Haemost. 2007, 5, 1149–1157. [Google Scholar] [CrossRef]
- Grève, E.; Moussata, D.; Gaudin, J.L.; Lapalus, M.-G.; Giraud, S.; Dupuis-Girod, S.; Calender, A.; Plauchu, H.; Saurin, J.-C. High diagnostic and clinical impact of small-bowel capsule endoscopy in patients with hereditary hemorrhagic telangiectasia with overt digestive bleeding and/or severe anemia. Gastrointest. Endosc. 2010, 71, 760–767. [Google Scholar] [CrossRef]
- Pierucci, P.; Lenato, G.M.; Suppressa, P.; Lastella, P.; Triggiani, V.; Valerio, R.; Comelli, M.; Salvante, D.; Stella, A.; Resta, N.; et al. A long diagnostic delay in patients with Hereditary Haemorrhagic Telangiectasia: A questionnaire-based retrospective study. Orphanet J. Rare Dis. 2012, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Mani, B.I.; Rubel, A.R.; Chauhdary, W.A.; Bashir, A.; Soe, Z.N.; Javed, N.; A Sharif, S.M.; Aye, M.T.H.; Chong, V.H.; Alam Sharif, S.M. Osler-Weber-Rendu syndrome. QJM Int. J. Med. 2020, 113, 586–587. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Buscarini, E.; Sabbà, C.; Mager, H.J.; Kjeldsen, A.D.; Pagella, F.; Sure, U.; Ugolini, S.; Toerring, P.; Suppressa, P.; et al. The European rare disease network for HHT frameworks for management of hereditary haemorrhagic telangiectasia in general and speciality care. Eur. J. Med. Genet. 2021, 65, 104370. [Google Scholar] [CrossRef]
- Begbie, M.E.; Wallace, G.M.F.; Shovlin, C.L. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): A view from the 21st century. Postgrad. Med. J. 2003, 79, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Gefen, A.M.; White, A.J. Asymptomatic pulmonary arteriovenous malformations in children with hereditary hemorrhagic telangiectasia. Pediatr. Pulmonol. 2017, 52, 1194–1197. [Google Scholar] [CrossRef]
- Giordano, P.; Francavilla, M.; Buonamico, P.; Suppressa, P.; Lastella, P.; Sangerardi, M.; Miniello, V.L.; Scardapane, A.; Lenato, G.M.; Sabbà, C.; et al. Hepatic angiodynamic profile in paediatric patients with hereditary haemorrhagic telangiectasia type 1 and type 2. Vasa 2017, 46, 195–202. [Google Scholar] [CrossRef]
- Hosman, A.E.; de Gussem, E.M.; Balemans, W.A.F.; Gauthier, A.; Westermann, C.J.J.; Snijder, R.J.; Post, M.C.; Mager, J.J. Screening children for pulmonary arteriovenous malformations: Evaluation of 18 years of experience. Pediatric Pulmonol. 2017, 52, 1206–1211. [Google Scholar] [CrossRef]
- Gonzalez, C.D.; Mcdonald, J.; Stevenson, D.A.; Whitehead, K.J.; Petersen, M.G.; Presson, A.P.; Ding, Q.; Wilson, K.F. Epistaxis in children and adolescents with hereditary hemorrhagic telangiectasia. Laryngoscope 2018, 128, 1714–1719. [Google Scholar] [CrossRef]
- Dingenouts, C.K.E.; Goumans, M.-J.; Bakker, W. Mononuclear cells and vascular repair in HHT. Front. Genet. 2015, 6, 114. [Google Scholar] [CrossRef] [Green Version]
- Al-Samkari, H.; Kasthuri, R.S.; Parambil, J.G.; Albitar, H.A.; Almodallal, Y.A.; Vázquez, C.; Serra, M.M.; Dupuis-Girod, S.; Wilsen, C.B.; McWilliams, J.P.; et al. An international, multicenter study of intravenous bevacizumab for bleeding in hereditary hemorrhagic telangiectasia: The InHIBIT-bleed study. Haematologica 2021, 106, 2161–2169. [Google Scholar] [CrossRef]
- Assar, O.S.; Friedman, C.M.; White, R.I. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991, 101, 977–980. [Google Scholar] [CrossRef]
- Robard, L.; Michel, J.; Prulière Escabasse, V.; Bequignon, E.; Vérillaud, B.; Malard, O.; Crampette, L.; Achache, M.; Lamrani, M.A.; Ardillon, L.; et al. Guidelines of the French Society of Otorhinolaryngology (SFORL) (short version). Specific treatment of epistaxis in Rendu-Osler-Weber disease. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2017, 134, 37–41. [Google Scholar] [CrossRef]
- Faughnan, M.E.; Mager, J.J.; Hetts, S.W.; Palda, V.A.; Lang-Robertson, K.; Buscarini, E.; Deslandres, E.; Kasthuri, R.S.; Lausman, A.; Poetker, D.; et al. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Ann. Intern. Med. 2020, 173, 989–1001. [Google Scholar] [CrossRef]
- Shields, H.M.; Shaffer, K.; O’farrell, R.P.; Travers, R.; Hayward, J.N.; Becker, L.S.; Lauwers, G.Y. Gastrointestinal manifestations of dermatologic disorders. Clin. Gastroenterol. Hepatol. 2007, 5, 1010–1017, quiz 1005–1006. [Google Scholar] [CrossRef]
- Notsu, T.; Adachi, K.; Mishiro, T.; Kishi, K.; Ishimura, N.; Ishihara, S. Prevalence of Angiodysplasia Detected in Upper Gastrointestinal Endoscopic Examinations. Cureus. 2021, 13, e14353. [Google Scholar] [CrossRef]
- Hammill, A.M.; Wusik, K.; Kasthuri, R.S. Hereditary hemorrhagic telangiectasia (HHT): A practical guide to management. Hematol. Am. Soc. Hematol. Educ. Program. 2021, 2021, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Mora-Luján, J.M.; Iriarte, A.; Alba, E.; Sánchez-Corral, M.Á.; Berrozpe, A.; Cerdà, P.; Cruellas, F.; Ribas, J.; Castellote, J.; Riera-Mestre, A. Gastrointestinal Bleeding in Patients with Hereditary Hemorrhagic Telangiectasia: Risk Factors and Endoscopic Findings. J. Clin. Med. 2019, 9, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.R.; Liu, H.; Wang, B.; Zhang, Y.H.; Xu, H.; Tang, X.L.; Li, H.M.; Zhao, S.Y. Case report of hereditary hemorrhagic telangiectasia in children and literature review. Zhonghua Er Ke Za Zhi Chin. J. Pediatrics 2020, 58, 674–678. [Google Scholar]
- Witkowska-Krawczak, E.; Zapolska, A.; Banaszkiewicz, A.; Kucharska, A. Recurrent gastrointestinal bleeding due to vascular malformations in a girl with Turner syndrome. Pediatr. Endocrinol. Diabetes Metab. 2021, 27, 222–226. [Google Scholar] [CrossRef]
- Hashmi, S.S.; Haider, N.; Malik, M.I. Hereditary Hemorrhagic Telangiectasia—Early Childhood Presentation with Hepatic Failure. J. Coll. Physicians Surg. Pak. 2018, 28, S195–S197. [Google Scholar] [PubMed]
- Gallitelli, M.; Pasculli, G.; Fiore, T.; Carella, A.; Sabbà, C. Emergencies in hereditary haemorrhagic telangiectasia. QJM Int. J. Med. 2006, 99, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Pahl, K.; Choudhury, A.; Kasthuri, R.S. Causes and severity of anemia in hereditary hemorrhagic telangiectasia. Blood 2016, 128, 3776. [Google Scholar] [CrossRef]
- Auerbach, M.; Adamson, J.W. How we diagnose and treat iron deficiency anemia. Am. J. Hematol. 2016, 91, 31–38. [Google Scholar] [CrossRef]
- Snook, J.; Bhala, N.; Beales, I.L.P.; Cannings, D.; Kightley, C.; Logan, R.P.; Pritchard, M.; Sidhu, R.; Surgenor, S.; Thomas, W.; et al. British Society of Gastroenterology guidelines for the management of iron deficiency anaemia in adults. Gut 2021, 70, 2030–2051. [Google Scholar] [CrossRef]
- Sonoda, K. Iron Deficiency Anemia: Guidelines from the American Gastroenterological Association. Am. Fam. Physician 2021, 104, 211–212. [Google Scholar]
- McDonagh, T.; Damy, T.; Doehner, W.; Lam, C.S.P.; Sindone, A.; van der Meer, P.; Cohen-Solal, A.; Kindermann, I.; Manito, N.; Pfister, O.; et al. Screening, diagnosis and treatment of iron deficiency in chronic heart failure: Putting the 2016 European Society of Cardiology heart failure guidelines into clinical practice. Eur. J. Heart Fail. 2018, 20, 1664–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Samkari, H. Hereditary hemorrhagic telangiectasia: Systemic therapies, guidelines, and an evolving standard of care. Blood 2021, 137, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Chetcuti Zammit, S.; Sanders, D.S.; McAlindon, M.E.; Sidhu, R. The Impact of Small Bowel Endoscopy in Patients with Hereditary Hemorrhagic Telangiectasia. Turk. J. Haematol. 2018, 35, 300–301. [Google Scholar] [PubMed]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Kritharis, A.; Rodriguez-Lopez, J.M.; Kuter, D.J. Systemic bevacizumab for the treatment of chronic bleeding in hereditary haemorrhagic telangiectasia. J. Intern. Med. 2019, 285, 223–231. [Google Scholar] [CrossRef]
- Al-Samkari, H. Systemic Bevacizumab for Hereditary Hemorrhagic Telangiectasia: Considerations from Observational Studies. Otolaryngol. Head Neck Surg. 2019, 160, 368. [Google Scholar] [CrossRef]
- Iyer, V.N.; Apala, D.R.; Pannu, B.S.; Kotecha, A.; Brinjikji, W.; Leise, M.D.; Kamath, P.S.; Misra, S.; Begna, K.H.; Cartin-Ceba, R.; et al. Intravenous Bevacizumab for Refractory Hereditary Hemorrhagic Telangiectasia-Related Epistaxis and Gastrointestinal Bleeding. Mayo Clin. Proc. 2018, 93, 155–166. [Google Scholar] [CrossRef]
- Guilhem, A.; Fargeton, A.E.; Simon, A.C.; Duffau, P.; Harle, J.R.; Lavigne, C.; Carette, M.F.; Bletry, O.; Kaminsky, P.; Leguy, V.; et al. Intra-venous bevacizumab in hereditary hemorrhagic telangiectasia (HHT): A retrospective study of 46 patients. PLoS ONE 2017, 12, e0188943. [Google Scholar] [CrossRef]
- Epperla, N.; Kleman, A.; Karafin, M.; Foy, P. Re-treatment versus extended treatment strategy of systemic bevacizumab in hereditary hemorrhagic telangiectasia: Which is better? Ann. Hematol. 2018, 97, 1727–1729. [Google Scholar] [CrossRef]
- Dupuis-Girod, S.; Ginon, I.; Saurin, J.-C.; Marion, D.; Guillot, E.; Decullier, E.; Roux, A.; Carette, M.-F.; Gilbert-Dussardier, B.; Hatron, P.-Y.; et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA 2012, 307, 948–955. [Google Scholar] [CrossRef] [Green Version]
- Lupu, A.; Stefanescu, C.; Treton, X.; Attar, A.; Corcos, O.; Bouhnik, Y. Bevacizumab as rescue treatment for severe recurrent gastrointestinal bleeding in hereditary hemorrhagic telangiectasia. J. Clin. Gastroenterol. 2013, 47, 256–257. [Google Scholar] [CrossRef]
- Ou, G.; Galorport, C.; Enns, R. Bevacizumab and gastrointestinal bleeding in hereditary hemorrhagic telangiectasia. World J. Gastrointest. Surg. 2016, 8, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, T.; Fialla, A.D.; Kjeldsen, J.; Kjeldsen, A.D. Does severe bleeding in HHT patients respond to intravenous bevacizumab? Review of the literature and case series. Rhinology 2019, 57, 242–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sargeant, I.R.; Loizou, L.A.; Rampton, D.; Tulloch, M.; Bown, S.G. Laser ablation of upper gastrointestinal vascular ectasias: Long term results. Gut 1993, 34, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J. Hepatol. 2007, 46, 499–507. [Google Scholar] [CrossRef]
- Buscarini, E.; Leandro, G.; Conte, D.; Danesino, C.; Daina, E.; Manfredi, G.; Lupinacci, G.; Brambilla, G.; Menozzi, F.; De Grazia, F.; et al. Natural history and outcome of hepatic vascular malformations in a large cohort of patients with hereditary hemorrhagic teleangiectasia. Dig. Dis. Sci. 2011, 56, 2166–2178. [Google Scholar] [CrossRef] [Green Version]
- Buscarini, E.; Plauchu, H.; Garcia Tsao, G.; White, R.I.; Sabbà, C.; Miller, F.; Saurin, J.C.; Pelage, J.P.; Lesca, G.; Marion, M.J.; et al. Liver involvement in hereditary hemorrhagic telangiectasia: Consensus recommendations. Liver Int. 2006, 26, 1040–1046. [Google Scholar] [CrossRef]
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Buscarini, E.; Chesnutt, M.S.; et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef]
- Kjeldsen, A.; Aagaard, K.S.; Tørring, P.M.; Möller, S.; Green, A. 20-year follow-up study of Danish HHT patients-survival and causes of death. Orphanet J. Rare Dis. 2016, 11, 157. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, T.; Cherif, H. Mutations in the ENG, ACVRL1, and SMAD4 genes and clinical manifestations of hereditary haemorrhagic telangiectasia: Experience from the Center for Osler’s Disease, Uppsala University Hospital. Ups. J. Med. Sci. 2018, 123, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Letteboer, T.G.W.; Mager, J.J.; Snijder, R.J.; Koeleman, B.P.C.; Lindhout, D.; van Amstel, J.K.P.; Westermann, C.J.J. Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J. Med. Genet. 2006, 43, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brakensiek, K.; Frye-Boukhriss, H.; Mälzer, M.; Abramowicz, M.; Bahr, M.J.; von Beckerath, N.; Bergmann, C.; Caselitz, M.; Holinski-Feder, E.; Muschke, P.; et al. Detection of a significant association between mutations in the ACVRL1 gene and hepatic involvement in German patients with hereditary haemorrhagic telangiectasia. Clin. Genet. 2008, 74, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Murray, E.; Taylor, J.; Hountras, P. A Case of High-Output Heart Failure. Chest 2022, 161, e23–e28. [Google Scholar] [CrossRef] [PubMed]
- Khoja, A.M.; Jalan, R.K.; Jain, D.L.; Kajale, O.V. Osler-Weber-Rendu disease: A rare cause of recurrent hemoptysis. Lung India 2016, 33, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Aubignat, M.; Salomon, A.; Chivot, C.; Delanghe, F.; Lecat, B.; Jeanjean, P.; Peltier, J. Abcès cérébral et maladie de Rendu-Osler-Weber: Pensez à rechercher des malformations artério-veineuses pulmonaires [Brain abscess and Osler-Weber-Rendu syndrome: Do not forget to look for pulmonary arteriovenous malformations]. Rev. Med. Interne 2020, 41, 776–779. (In French) [Google Scholar] [CrossRef]
- De Gussem, E.M.; Kroon, S.; Hosman, A.E.; Kelder, J.C.; Post, M.C.; Snijder, R.J.; Mager, J. Hereditary Hemorrhagic Telangiectasia (HHT) and Survival: The Importance of Systematic Screening and Treatment in HHT Centers of Excellence. J. Clin. Med. 2020, 9, 3581. [Google Scholar] [CrossRef]
- Cottin, V.; Plauchu, H.; Bayle, J.Y.; Barthelet, M.; Revel, D.; Cordier, J.F. Pulmonary arteriovenous malformations in patients with hereditary hemorrhagic telangiectasia. Am. J. Respir. Crit. Care Med. 2004, 169, 994–1000. [Google Scholar] [CrossRef] [Green Version]
- Brinjikji, W.; Nasr, D.M.; Cloft, H.J.; Iyer, V.N.; Lanzino, G. Spinal arteriovenous fistulae in patients with hereditary hemorrhagic telangiectasia: A case report and systematic review of the literature. Interv. Neuroradiol. 2016, 22, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Shovlin, C.L.; Millar, C.M.; Droege, F.; Kjeldsen, A.; Manfredi, G.; Suppressa, P.; Ugolini, S.; Coote, N.; Fialla, A.D.; Geisthoff, U.; et al. Safety of direct oral anticoagulants in patients with hereditary hemorrhagic telangiectasia. Orphanet J. Rare Dis. 2019, 14, 210. [Google Scholar] [CrossRef]
- Dittus, C.; Streiff, M.; Ansell, J. Bleeding and clotting in hereditary hemorrhagic telangiectasia. World J. Clin. Cases 2015, 3, 330–337. [Google Scholar] [CrossRef]
- Orizaga-Y-Quiroga, T.L.; Villarreal-Martínez, A.; Jaramillo-Moreno, G.; Ocampo-Candiani, J. Osler-Weber-Rendu Syndrome in Relation to Dermatology. Actas Dermosifiliogr. (Engl. Ed.) 2019, 110, 526–532. (In English) [Google Scholar] [CrossRef] [PubMed]
- Mikołajczyk-Solińska, M.; Leończyk, K.; Brzezina, A.; Rossa, S.; Kasznicki, J. Life-threatening Anaemia in Patient with Hereditary Haemorrhagic Telangiectasia (Rendu-Osler-Weber Syndrome). Open Med. 2020, 15, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Arai, N.; Akiyama, T. A questionnaire-based survey to evaluate and improve the current HHT medical and social condition in Japan. Surg. Neurol. Int. 2020, 11, 323. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Symptoms | |
|---|---|
| 1 | Epistaxis—spontaneous and recurrent |
| 2 | Telangiectasias: multiple and characteristic sites (lips, mouth, fingers, nose) |
| 3 | Visceral/gastrointestinal telangiectasias, pulmonary AVMs 1, hepatic AVMs, cerebral AVMs and spinal AVMs |
| 4 | Family history: one first-degree relative |
| Laboratory Test | Result | Normal Range |
|---|---|---|
| Hemoglobin | 9.0 g/dL | 11.5–16 g/dL |
| Hematocrit | 29.4% | 37–47% |
| Red blood count | 3.91 T/L | 3.5–5.2 T/L |
| Platelets | 266/mm3 | 150–400/mm3 |
| Leukocytes | 3420/mm3 | 4000–10,000/mm3 |
| Lymphocytes | 11.8% | 25–40% |
| Neutrophils | 65% | 50–62% |
| Alanine aminotransferase | 14 U/L | <33 U/L |
| Asparate aminotransferase | 16 U/L | <32 U/L |
| Total bilirubin | 0.7 mg/dL | 0.2–1.2 mg/dL |
| Creatinine | 0.7 mg/dL | 0.5–1.1 mg/dL |
| C-reactive protein | 7.1 mg/L | 0–5 mg/L |
| D-dimer | 315 ng/mL | 0–500 ng/mL |
| Sodium | 138 mmol/L | 136–145 mmol/L |
| Potassium | 4.0 mmol/L | 3.5–5.1 mmol/L |
| Iron | 10 ug/dL | 50–170 ug/dL |
| Ferritin | 7 ng/mL | 10–291 ng/mL |
| Vitamin B12 | 392 pg/mL | 211–911 pg/mL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jargielo, A.; Rycyk, A.; Kasztelan-Szczerbinska, B.; Cichoz-Lach, H. A Rare Case of Upper Gastrointestinal Bleeding: Osler-Weber-Rendu Syndrome. Medicina 2022, 58, 333. https://doi.org/10.3390/medicina58030333
Jargielo A, Rycyk A, Kasztelan-Szczerbinska B, Cichoz-Lach H. A Rare Case of Upper Gastrointestinal Bleeding: Osler-Weber-Rendu Syndrome. Medicina. 2022; 58(3):333. https://doi.org/10.3390/medicina58030333
Chicago/Turabian StyleJargielo, Anna, Anna Rycyk, Beata Kasztelan-Szczerbinska, and Halina Cichoz-Lach. 2022. "A Rare Case of Upper Gastrointestinal Bleeding: Osler-Weber-Rendu Syndrome" Medicina 58, no. 3: 333. https://doi.org/10.3390/medicina58030333
APA StyleJargielo, A., Rycyk, A., Kasztelan-Szczerbinska, B., & Cichoz-Lach, H. (2022). A Rare Case of Upper Gastrointestinal Bleeding: Osler-Weber-Rendu Syndrome. Medicina, 58(3), 333. https://doi.org/10.3390/medicina58030333