
Oleuropein Prevents Neuronal Death, Mitigates Mitochondrial Superoxide Production and Modulates Autophagy in a Dopaminergic Cellular Model

Abstract
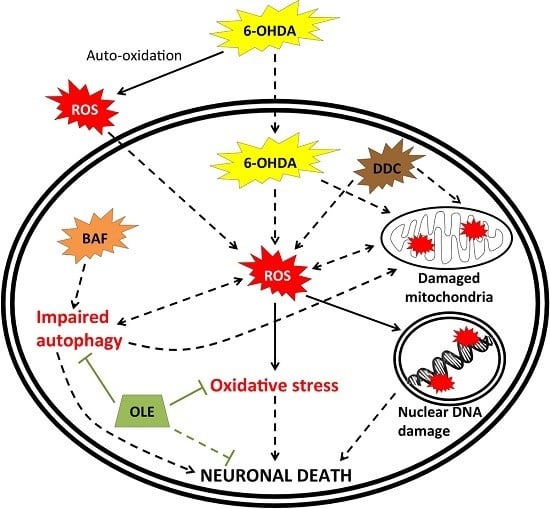
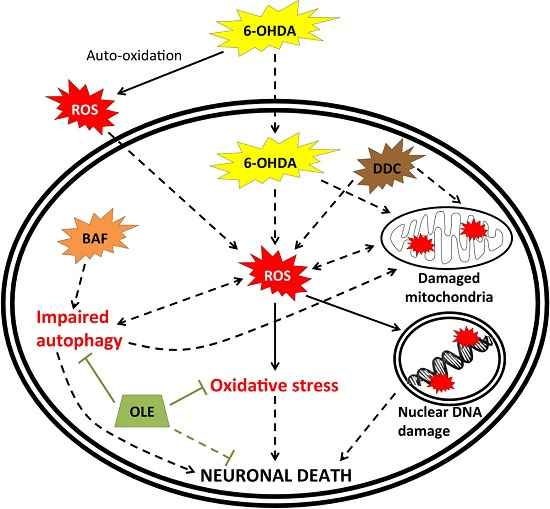
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
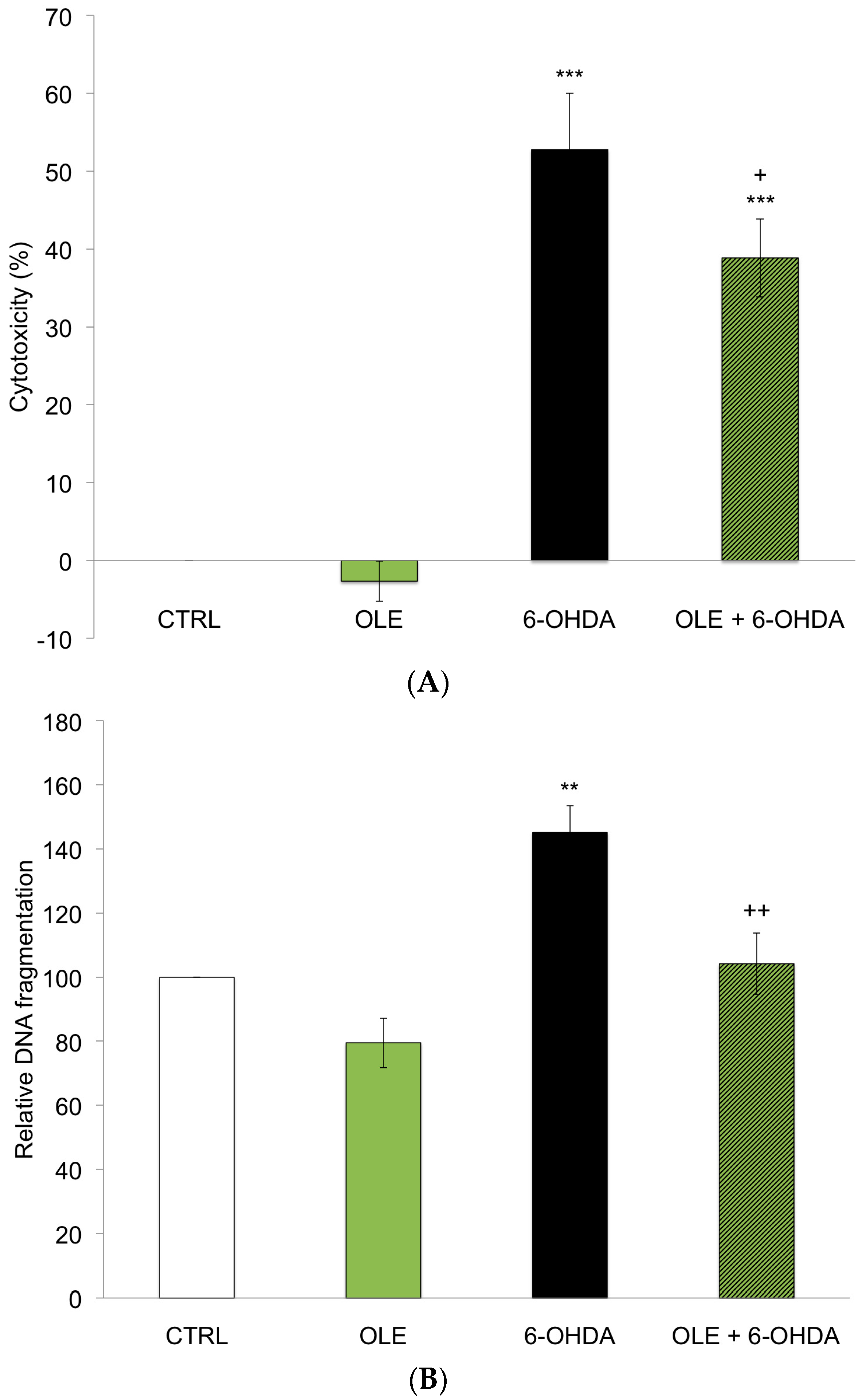
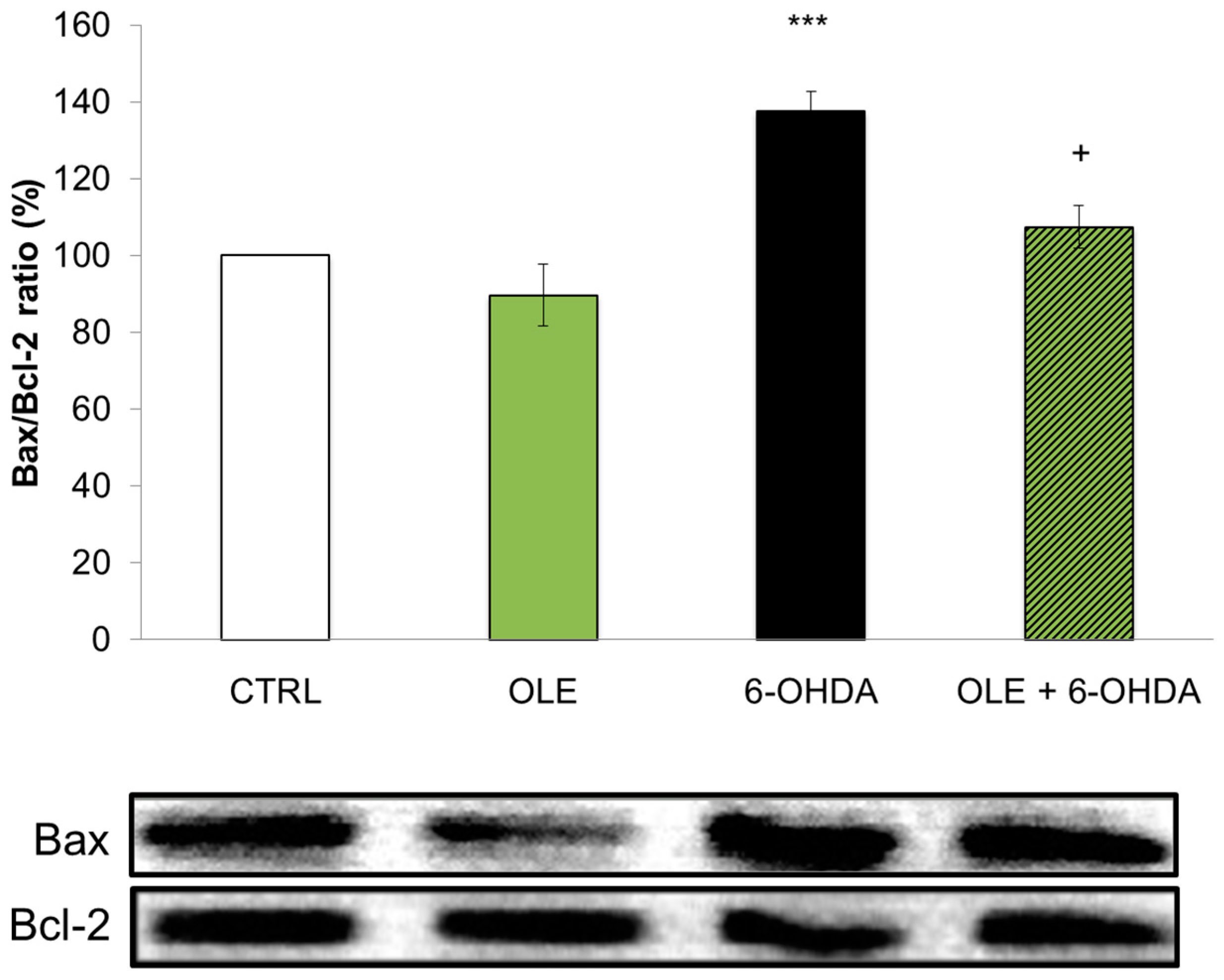
2.1. OLE Prevents 6-OHDA-Induced Neuronal Death
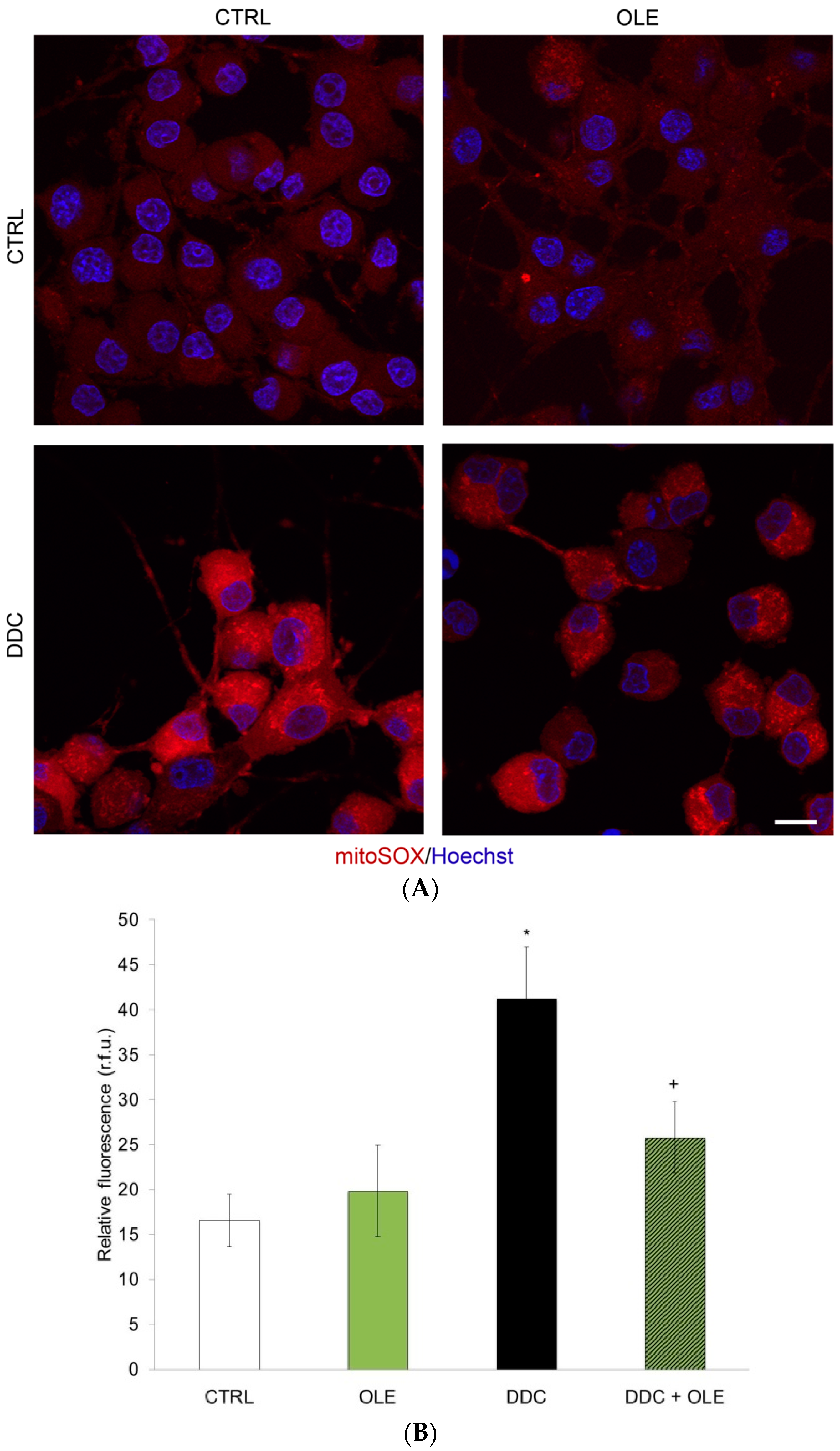
2.2. OLE Mitigates Mitochondrial Superoxide Production
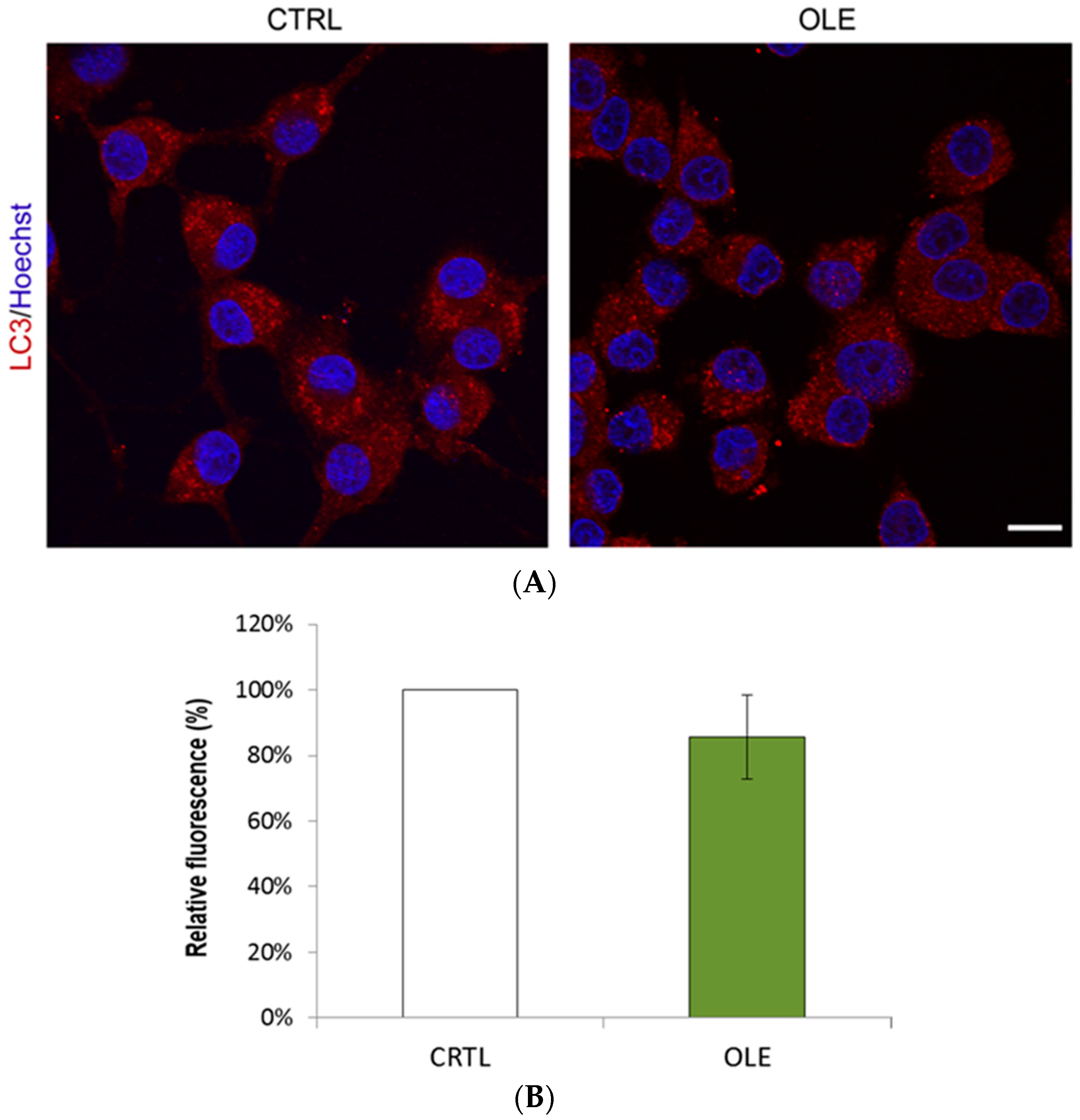
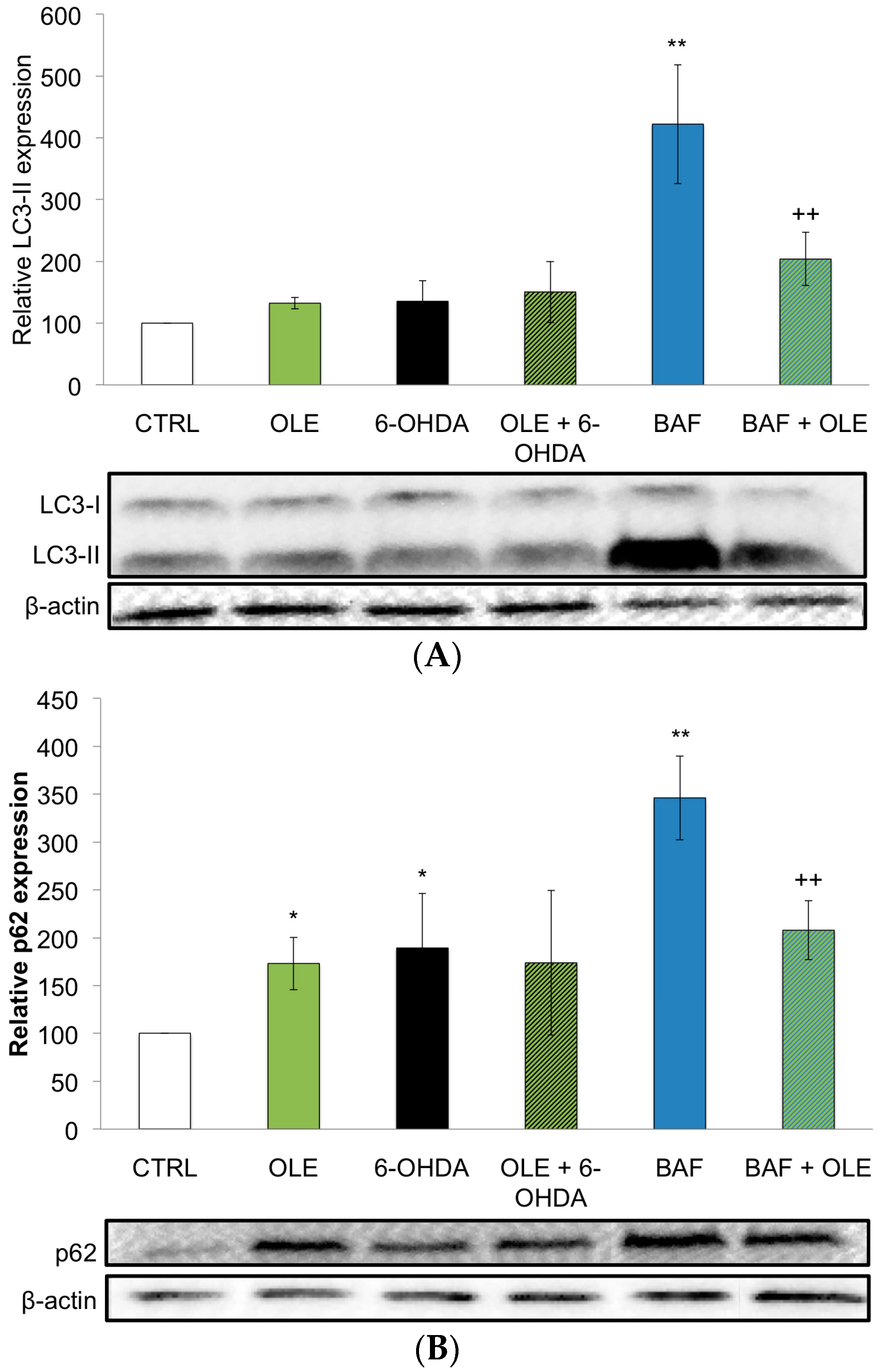
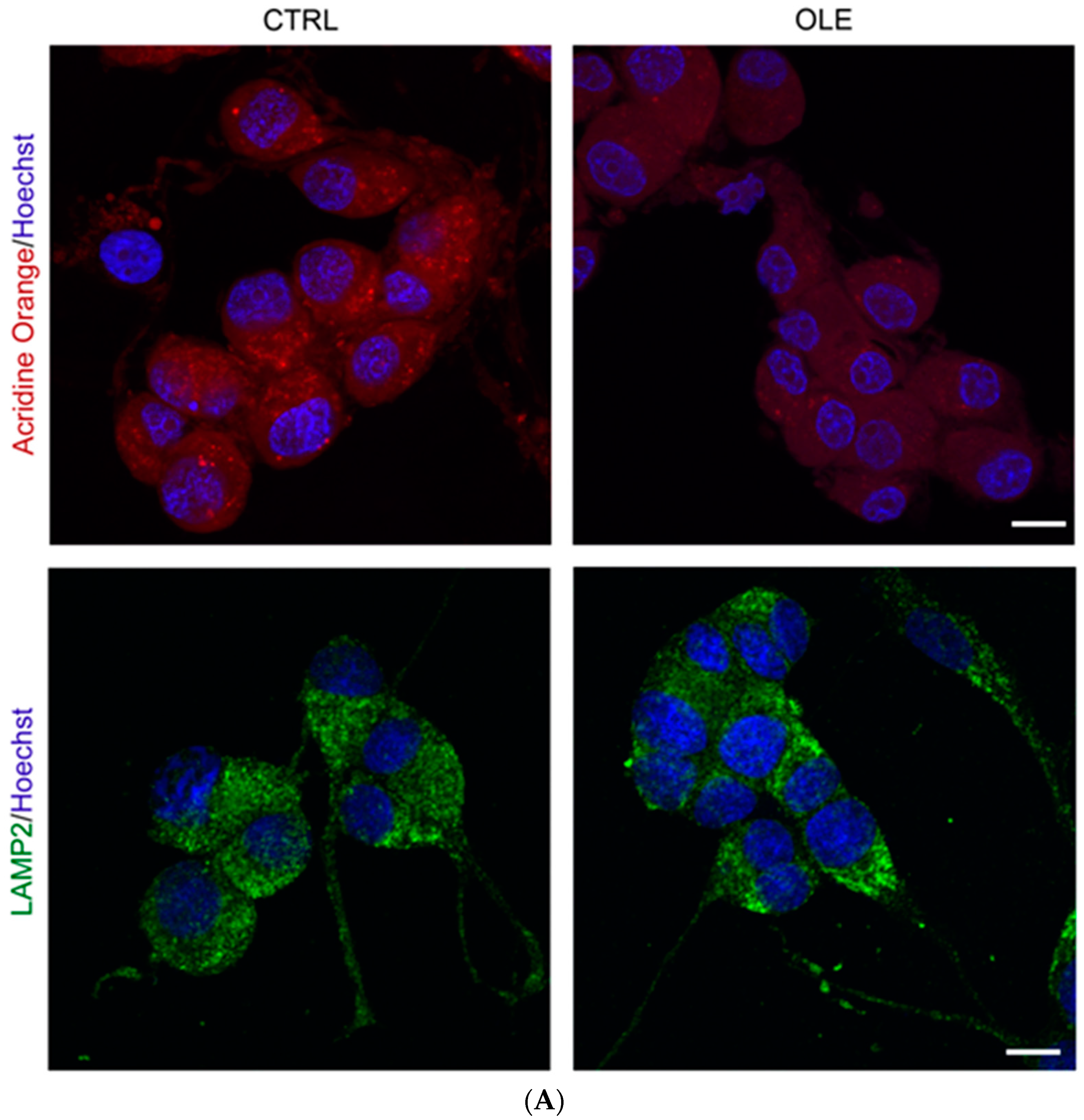
2.3. OLE Modulates Autophagy
3. Discussion
4. Materials and Methods
4.1. Drugs and Chemicals
4.2. Cell Culture and Treatments
4.3. Cytotoxicity Measurements
4.4. Apoptosis-Specific DNA Denaturation
4.5. Detection of Mitochondrial Superoxide Anion
4.6. Detection of Acidic Vesicles by Acridine Orange
4.7. Immunofluorescence Microscopy
4.8. Western Blotting Assays
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Huenchuguala, S.; Paris, I.; Segura-Aguilar, J. Dopamine oxidation and autophagy. Parkinson’s Dis. 2012. [Google Scholar] [CrossRef]
- Xiang, W.; Schlachetzki, J.C.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schäffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.M. Oxidative stress-induced posttranslational modifications of alpha-synuclein: specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013, 54, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Kleinknecht, A.; Popova, B.; Lázaro, D.F.; Pinho, R.; Valerius, O.; Outeiro, T.F.; Braus, G.H. C-terminal tyrosine residue modifications modulate the protective phosphorylation of serine 129 of α-synuclein in a yeast model of Parkinson’s disease. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [PubMed]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediators Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed]
- Tsang, A.H.; Chung, K.K. Oxidative and nitrosative stress in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Schuman, E.M. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunctions. Nat. Rev. Neurosci. 2008, 9, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Germain, M.; Kelly, M.A.; Slack, R.S. Reactive oxygen species: Stuck in the middle of neurodegeneration. J. Alzheimers Dis. 2010, 20, S357–S367. [Google Scholar] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Perluigi, M.; Allan Butterfield, D. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [PubMed]
- Soper, J.H.; Kehm, V.; Burd, C.G.; Bankaitis, V.A.; Lee, V.M. Aggregation of alpha-synuclein in S. cerevisiae is associated with defects in endosomal trafficking and phospholipid biosynthesis. J. Mol. Neurosci. 2011, 43, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Beal, M.F.; Thomas, B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010, 33, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Son, J.H.; Shim, J.H.; Kim, K.H.; Ha, J.Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Kondo, S.; Le, W.; Jankovic, J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 2008, 131, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Redmann, M.; Darley-Usmar, V.; Zhang, J. The role of autophagy, mitophagy and lysosomal functions in modulating bioenergetics and survival in the context of redox and proteotoxic damage: Implications for neurodegenerative diseases. Aging Dis. 2016, 7, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Yu, J.T.; Tan, M.S.; Jiang, T.; Zhu, X.C.; Tan, L. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol. Aging 2014, 35, 941–957. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Jiang, T.; Guo, J.; Liu, Y.; Cui, G.; Gu, L.; Su, L.; Zhang, Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, J.; Ni, W.; Shen, Y.W.; Zhang, X.P. Peroxisome proliferator-activated receptor-γ agonist 15d-prostaglandin J2 mediates neuronal autophagy after cerebral ischemia-reperfusion injury. PLoS ONE 2013, 8, e55080. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Raymick, J.; Imam, S. Neuroprotective and Therapeutic Strategies against Parkinson’s Disease: Recent Perspectives. Int. J. Mol. Sci. 2016, 17, 904. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Virmani, A.; Pinto, L.; Binienda, Z.; Syed, A. Neuroprotection by what you eat. Mol. Neurobiol. 2013, 48, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.; Calon, F.; Cicchetti, F. Impact of omega-3 fatty acids in Parkinson’s disease. Ageing Res. 2011, 10, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Koppula, S.; Kumar, H.; More, S.V.; Lim, H.W.; Hong, S.M.; Choi, D.K. Recent updates in redox regulation and free radical scavenging effects by herbal products in experimental models of Parkinson’s disease. Molecules 2012, 17, 11391–11420. [Google Scholar] [CrossRef] [PubMed]
- Hang, L.; Basil, A.H.; Lim, K.L. Nutraceuticals in Parkinson’s Disease. Neuromol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Rodriguez-Mateos, A.; Corona, G.; Oruna-Concha, M.J.; Spencer, J.P. Polyphenols and human health: Prevention of disease and mechanisms of action. Nutrients 2010, 2, 1106–1131. [Google Scholar] [CrossRef] [PubMed]
- Rigacci, S.; Stefani, M. Nutraceutical properties of olive oil polyphenols: An itinerary from cultured cells through animal models to humans. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Umeno, A.; Horie, M.; Murotomi, K.; Nakajima, Y.; Yoshida, Y. Antioxidative and Antidiabetic Effects of Natural Polyphenols and Isoflavones. Molecules 2016, 21, 708. [Google Scholar] [CrossRef] [PubMed]
- Attard, E.; Martinoli, M.G. Cucurbitacin E, An experimental lead triterpenoid with anticancer, immunomodulatory and novel effects against degenerative diseases. Curr. Top. Med. Chem. 2015, 15, 1708–1713. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.; Nabavi, S.F.; Daglia, M.; Nabavi, S.M.; Martinoli, M.G. Epigallocatechin-3-gallate, a promising molecule for Parkinson’s Disease? Rejuvenation Res. 2015, 18, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.; Martinoli, M.G. Resveratrol as a protective molecule for neuroinflammation: A review of mechanisms. Curr. Pharm. Biotechnol. 2014, 15, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, H.; Albutti, A.S.; Aly, S.M. Therapeutics role of olive fruits/oil in the prevention of diseases via modulation of anti-oxidant, anti-tumour and genetic activity. Int. J. Clin. Exp. Med. 2014, 7, 799–808. [Google Scholar] [PubMed]
- Efentakis, P.; Iliodromitis, E.K.; Mikros, E.; Papachristodoulou, A.; Dagres, N.; Skaltsounis, A.L.; Andreadou, I. Effects of the olive tree leaf constituents on myocardial oxidative damage and atherosclerosis. Planta Med. 2015, 81, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Iliodromitis, E.K.; Mikros, E.; Constantinou, M.; Agalias, A.; Magiatis, P.; Skaltsounis, A.L.; Kamber, E.; Tsantili-Kakoulidou, A.; Kremastinos, D.T. The olive constituent oleuropein exhibits antiischemic, antioxidative, and hypolipidemic effects in anesthetized rabbits. J. Nutr. 2006, 136, 2213–2219. [Google Scholar] [PubMed]
- Sarbishegi, M.; Mehraein, F.; Soleimani, M. Antioxidant role of oleuropein on midbrain and dopaminergic neurons of substantia nigra in aged rats. Iran. Biomed. J. 2014, 18, 16–22. [Google Scholar] [PubMed]
- Pasban-Aliabadi, H.; Esmaeili-Mahani, S.; Sheibani, V.; Abbasnejad, M.; Mehdizadeh, A.; Yaghoobi, M.M. Inhibition of 6-hydroxydopamine-induced PC12 cell apoptosis by olive (Olea europaea L.) leaf extract is performed by its main component oleuropein. Rejuvenation Res. 2013, 16, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Pantano, D.; Luccarini, I.; Nardiello, P.; Servili, M.; Stefani, M.; Casamenti, F. Oleuropein aglycone and polyphenols from olive mill wastewater ameliorate cognitive deficits and neuropathology. Br. J. Clin. Pharmacol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Casamenti, F.; Grossi, C.; Rigacci, S.; Pantano, D.; Luccarini, I.; Stefani, M. Oleuropein aglycone: A possible drug against degenerative conditions: In vivo evidence of its effectiveness against Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 679–688. [Google Scholar] [PubMed]
- Rigacci, S.; Miceli, C.; Nediani, C.; Berti, A.; Cascella, R.; Pantano, D.; Nardiello, P.; Luccarini, I.; Casamenti, F.; Stefani, M. Oleuropein aglycone induces autophagy via the AMPK/mTOR signalling pathway: A mechanistic insight. Oncotarget 2015, 6, 35344–35357. [Google Scholar] [PubMed]
- Van Cauwenberghe, C.; Vandendriessche, C.; Libert, C.; Vandenbroucke, R.E. Caloric restriction: Beneficial effects on brain aging and Alzheimer’s disease. Mamm. Genome 2016, 27, 300–319. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.; Bournival, J.; Zottig, X.; Martinoli, M.G. Resveratrol protects DAergic PC12 cells from high glucose-induced oxidative stress and apoptosis: Effect on p53 and GRP75 localization. Neurotox. Res. 2014, 25, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Arel-Dubeau, A.M.; Longpre, F.; Bournival, J.; Tremblay, C.; Demers-Lamarche, J.; Hasvoka, P.; Attard, E.; Germain, M.; Martinoli, M.G. Cucurbitacin E has neuroprotective properties and autophagic modulating activities on dopaminergic neurons. Oxid. Med. Cell. Longev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Carange, J.; Longpré, F.; Daoust, B.; Martinoli, M.G. 24-Epibrassinolide, a phytosterol from the brassinosteroid family, protects dopaminergic cells against MPP+-induced oxidative stress and apoptosis. J. Toxicol. 2011, 392859. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; Bamber, B.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Iavicoli, I.; di Paola, R.; Koverech, A.; Cuzzocrea, S.; Rizzarelli, E.; Calabrese, E.J. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim. Biophys. Acta 2012, 1822, 753–783. [Google Scholar] [CrossRef] [PubMed]
- Nile, S.H.; Park, S.W. Edible berries: bioactive components and their effect on human health. Nutrition 2014, 30, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.L.; Silva, V.D.; Dos Santos Souza, C.; Santos, C.C.; Paris, I.; Muñoz, P.; Segura-Aguilar, J. Impact of Plant-Derived Flavonoids on Neurodegenerative Diseases. Neurotox. Res. 2016, 30, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Reglodi, D.; Renaud, J.; Tamas, A.; Tizabi, Y.; Socías, S.B.; del-Bel, E.; Raisman-Vozari, R. Novel tactics for neuroprotection in Parkinson’s disease: Role of antibiotics, polyphenols and neuropeptides. Prog. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Thorstensen, E.B.; Derraik, J.G.; Henderson, H.V.; Hofman, P.L.; Cutfield, W.S. Human absorption and metabolism of oleuropein and hydroxytyrosol ingested as olive (Olea europaea L.) leaf extract. Mol. Nutr. Food Res. 2013, 57, 2079–2085. [Google Scholar] [CrossRef]
- García-Villalba, R.; Larrosa, M.; Possemiers, S.; Tomás-Barberán, F.A.; Espín, J.C. Bioavailability of phenolics from an oleuropein-rich olive (Olea europaea) leaf extract and its acute effect on plasma antioxidant status: comparison between pre- and postmenopausal women. Eur. J. Nutr. 2014, 53, 1015–1027. [Google Scholar] [CrossRef]
- Gélinas, S.; Martinoli, M.G. Neuroprotective effect of estradiol and phytoestrogens on MPP+-induced cytotoxicity in neuronal PC12 cells. J. Neurosci. Res. 2002, 70, 90–96. [Google Scholar] [CrossRef]
- Kadota, T.; Yamaai, T.; Saito, Y.; Akita, Y.; Kawashima, S.; Moroi, K.; Inagaki, N.; Kadota, N. Expression of dopamine transporter at the tips of growing neurites of PC12 cells. J. Histochem. Cytochem. 1996, 44, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, J.; Mor, G.; Naftolin, F. Raloxifene induces neurite outgrowth in estrogen receptor positive PC12 cells. Menopause 1998, 5, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Chiasson, K.; Lahaie-Collins, V.; Bournival, J.; Delapierre, B.; Gélinas, S.; Martinoli, M.G. Oxidative stress and 17 alpha and 17 beta estradiol modulate neurofilaments differently. J. Mol. Neurosci. 2006, 30, 297–309. [Google Scholar] [CrossRef]
- Bournival, J.; Francoeur, M.A.; Renaud, J.; Martinoli, M.G. Quercetin and sesamin protect neuronal PC12 cells from high-glucose-induced oxidation, nitrosative stress and apoptosis. Rejuvenation Res. 2012, 15, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Blasina, M.F.; Vaamonde, L.; Morquio, A.; Echeverry, C.; Arredondo, F.; Dajas, F. Differentiation induced by Achyrocline satureioides (Lam) infusion in PC12 cells. Phytother. Res. 2009, 23, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- El Omri, A.; Han, J.; Yamada, P.; Kawada, K.; Ben Abdrabbah, M.; Isoda, H. Rosmarinus officinalis polyphenols activate cholinergic activities in PC12 cells through phosphorylation of ERK1/2. J. Ethnopharmacol. 2010, 131, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olatunji, O.J.; Feng, Y.; Olatunji, O.O.; Tang, J.; Ouyang, Z.; Su, Z. Cordycepin protects PC12 cells against 6-hydroxydopamine induced neurotoxicity via its antioxidant properties. Biomed. Pharmacother. 2016, 81, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Bournival, J.; Quessy, P.; Martinoli, M.G. Protective effects of resveratrol and quercetin against MPP+-induced oxidative stress act by modulating markers of apoptotic death in dopaminergic neurons. Cell. Mol. Neurobiol. 2009, 29, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gao, L.; Song, Y.; Qin, Z.-H.; Liang, Z. Activated cathepsin L is associated with the switch from autophagy to apoptotic death of SH-SY5Y cells exposed to 6-hydroxydopamine. Biochem. Biophys. Res. Commun. 2016, 470, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Beertsen, W.; Eskelinen, E.L. LAMP2, a control step for phagosome and autophagosome maturation. Autophagy 2008, 4, 510–512. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Joven, J.; Aragonès, G.; Barrajón-Catalán, E.; Beltrán-Debón, R.; Borrás-Linares, I.; Camps, J.; Corominas-Faja, B.; Cufí, S.; Fernández-Arroyo, S.; et al. Xenohormetic and anti-aging activity of secoiridoid polyphenols present in extra virgin olive oil: a new family of gerosuppressant agents. Cell Cycle 2013, 12, 555–578. [Google Scholar] [CrossRef] [PubMed]
- Dattilo, S.; Mancuso, C.; Koverech, G.; Di Mauro, P.; Ontario, M.L.; Petralia, C.C.; Petralia, A.; Maiolino, L.; Serra, A.; Calabrese, E.J.; et al. Heat shock proteins and hormesis in the diagnosis and treatment of neurodegenerative diseases. Immun. Ageing 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Kikusato, M.; Muroi, H.; Uwabe, Y.; Furukama, K.; Toyomizu, M. Oleuropein induces mitochondrial biogenesis and decreases reactive oxygen species generation in cultured avian muscle cells, possibly via an up-regulation of peroxisome proliferator-activated receptor γ coactivator-1α. Anim. Sci. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Mehta, S.L.; Kaimal, B.; Lyons, K.; Dempsey, R.J.; Vemuganti, R. Poststroke induction of α-synuclein mediates ischemic brain damage. J. Neurosci. 2016, 36, 7055–7065. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.Y.; Ye, Q.; Huang, H.J.; Xia, N.G.; Chen, Y.Y.; Zhang, Y.; Qu, Q.M. Salidroside protects cortical neurons against glutamate-induced cytotoxicity by inhibiting autophagy. Mol. Cell. Biochem. 2016, 419, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Wong, P.M.; Gammoh, N.; Jiang, X. Disctinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell 2011, 42, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Lohmann-Matthes, M.L. A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods 1998, 115, 61–69. [Google Scholar] [CrossRef]
- Frankfurt, O.S.; Krishan, A. Identification of apoptotic cells by formamide-induced DNA denaturation in condensed chromatin. J. Histochem. Cytochem. 2001, 49, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Tsai, S.-H.; Tseng, M.T.; Peng, S.-F.; Kuo, C.-S.; Lin, M.-W.; Hsu, Y.-M.; Lee, M.-R.; Amagaya, S.; Huang, W.-W.; et al. AKT serine/threonine protein kinase modulates baicalin-triggered autophagy in human bladder cancer T24 cells. Int. J. Oncol. 2013, 42, 993–1000. [Google Scholar] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Achour, I.; Arel-Dubeau, A.-M.; Renaud, J.; Legrand, M.; Attard, E.; Germain, M.; Martinoli, M.-G. Oleuropein Prevents Neuronal Death, Mitigates Mitochondrial Superoxide Production and Modulates Autophagy in a Dopaminergic Cellular Model. Int. J. Mol. Sci. 2016, 17, 1293. https://doi.org/10.3390/ijms17081293
Achour I, Arel-Dubeau A-M, Renaud J, Legrand M, Attard E, Germain M, Martinoli M-G. Oleuropein Prevents Neuronal Death, Mitigates Mitochondrial Superoxide Production and Modulates Autophagy in a Dopaminergic Cellular Model. International Journal of Molecular Sciences. 2016; 17(8):1293. https://doi.org/10.3390/ijms17081293
Chicago/Turabian StyleAchour, Imène, Anne-Marie Arel-Dubeau, Justine Renaud, Manon Legrand, Everaldo Attard, Marc Germain, and Maria-Grazia Martinoli. 2016. "Oleuropein Prevents Neuronal Death, Mitigates Mitochondrial Superoxide Production and Modulates Autophagy in a Dopaminergic Cellular Model" International Journal of Molecular Sciences 17, no. 8: 1293. https://doi.org/10.3390/ijms17081293
APA StyleAchour, I., Arel-Dubeau, A.-M., Renaud, J., Legrand, M., Attard, E., Germain, M., & Martinoli, M.-G. (2016). Oleuropein Prevents Neuronal Death, Mitigates Mitochondrial Superoxide Production and Modulates Autophagy in a Dopaminergic Cellular Model. International Journal of Molecular Sciences, 17(8), 1293. https://doi.org/10.3390/ijms17081293