
Hypokalemia in Diabetes Mellitus Setting


Abstract
:1. Introduction
2. Hypokalemia and DM
2.1. Definition, Prevalence, and Importance
2.2. Main Causes of Hypokalemia in Individuals with DM
2.2.1. Transcellular Shifts Caused by Drugs
Insulin
Beta-2 Agonists
Other Agents
2.2.2. Abnormal Potassium Losses Caused by Drugs
Diuretics
Glucocorticoids and Mineralocorticoids
Antibiotics
Oral Anti-Diabetics and Potassium
2.2.3. Induced Gastrointestinal Losses
2.2.4. Nondrug-Induced Transcellular Shifts
2.2.5. Nondrug-Induced Potassium Losses
2.2.6. Genetic Causes of Hypokalemia
- (a)
- increased K+ shift, including familial hypokalemia periodic paralysis or FPP (an autosomal dominant disorder caused by mutations on ion channel genes encoding the dihydropyridine-sensitive voltage-gated Ca2+ channel α1-subunit (CACNA1S) [FPP type I] and tetrodotoxin-sensitive voltage-gated Na+ channel α-subunit [SCN4A] [FPP type II] of skeletal muscle) and Andersen-Tawil syndrome (associated with mutations in the gene (KCNJ2) encoding a pore-forming subunit of the inward rectifier K+ channel protein, Kir2.1, which is expressed in heart and skeletal muscles);
- (b)
- defects in the intestinal tract (characterized by a mutation in the downregulated in adenoma (DRA) gene encoding a Cl−-OH− (HCO3−) exchanger expressed in the apical membranes of the colon and ileum, which leads to watery diarrhea, hypochloremic metabolic acidosis, and hypokalemia);
- (c)
- defects in exocrine glands (cystic fibrosis is associated with defective chloride reabsorption by the dysfunctional CFTR [cystic fibrosis transmembrane regulator] in the sweat ducts which is responsible for excessive Cl− and Na+ loss in sweat, leading to ECF volume depletion and, ultimately, to secondary hyperaldosteronism). To note, in familial hypokalemia periodic paralysis, attacks can be induced not only by rest after exercise, carbohydrate-rich meals, or exposure to cold but also by the administration of glucose or insulin or glucocorticoid, which can put diabetic patients at greater risk.
- (a)
- increased urine flow rate to cortical collecting ducts;
- (b)
- increased K+ concentration in the cortical collecting ducts.
2.3. DM-Related Acute Complications: Diabetic Ketoacidosis (DKA), Hyperglycemic Hyperosmolar State (HHS), and Euglycemic Diabetic Ketoacidosis (EDKA)
2.3.1. Diabetic Ketoacidosis
2.3.2. Hyperglicemic Hyperosmolar State
2.3.3. Euglycemic Diabetic Ketoacidosis
3. Symptoms, Exams, and Diagnosis of Hypokalemia
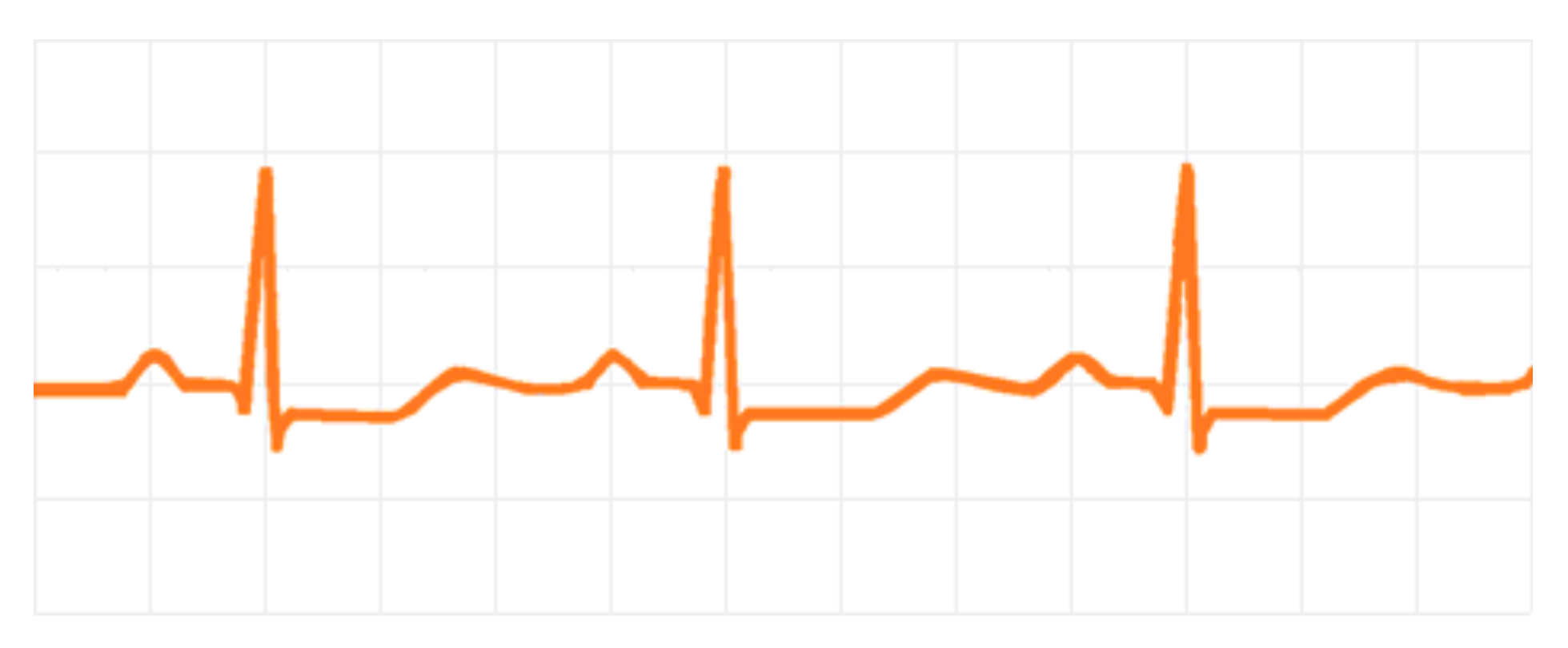
3.1. Cardiovascular Effects
3.2. Muscular Effects
3.3. Kidney Effects
3.4. Hormonal Effects
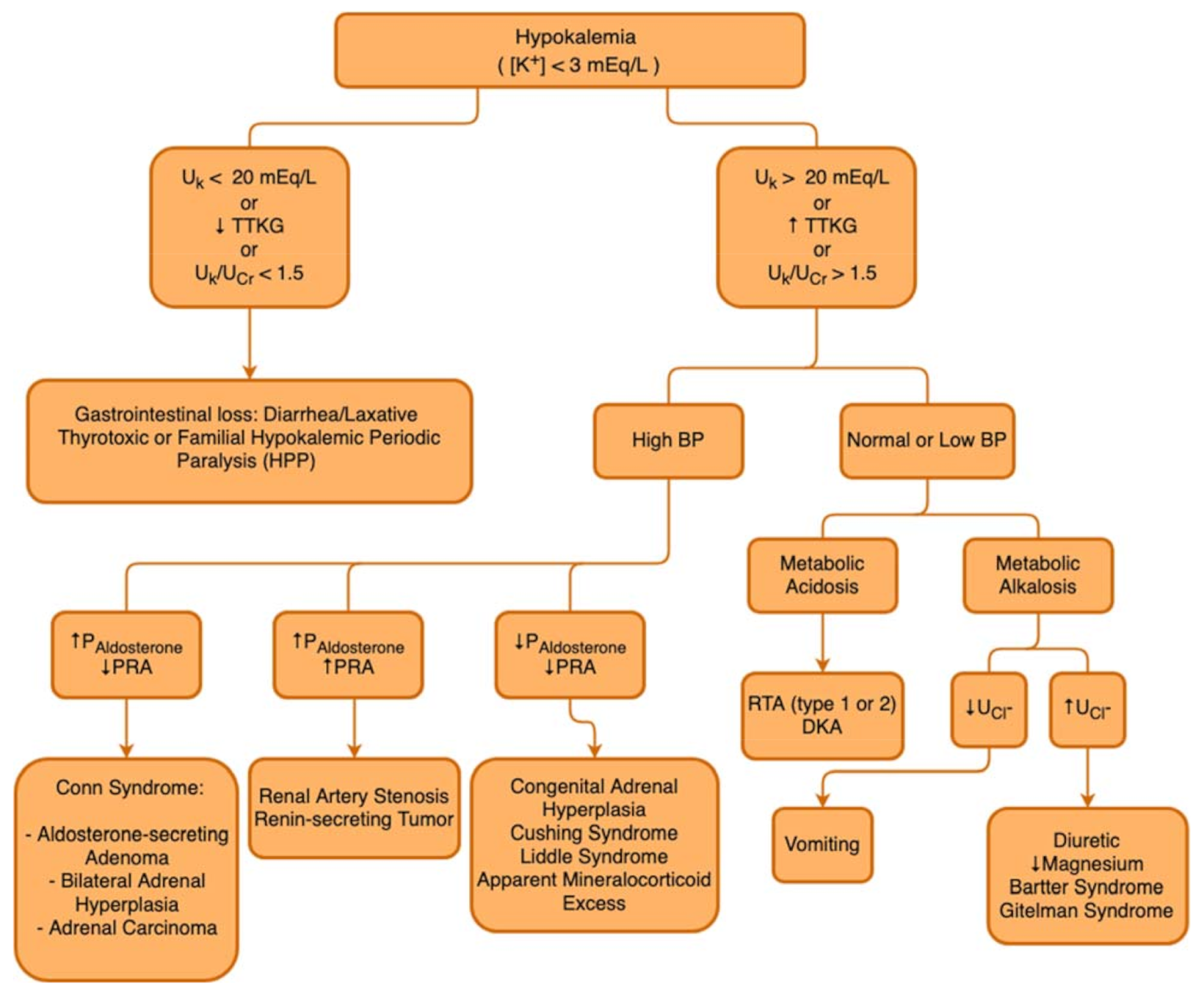
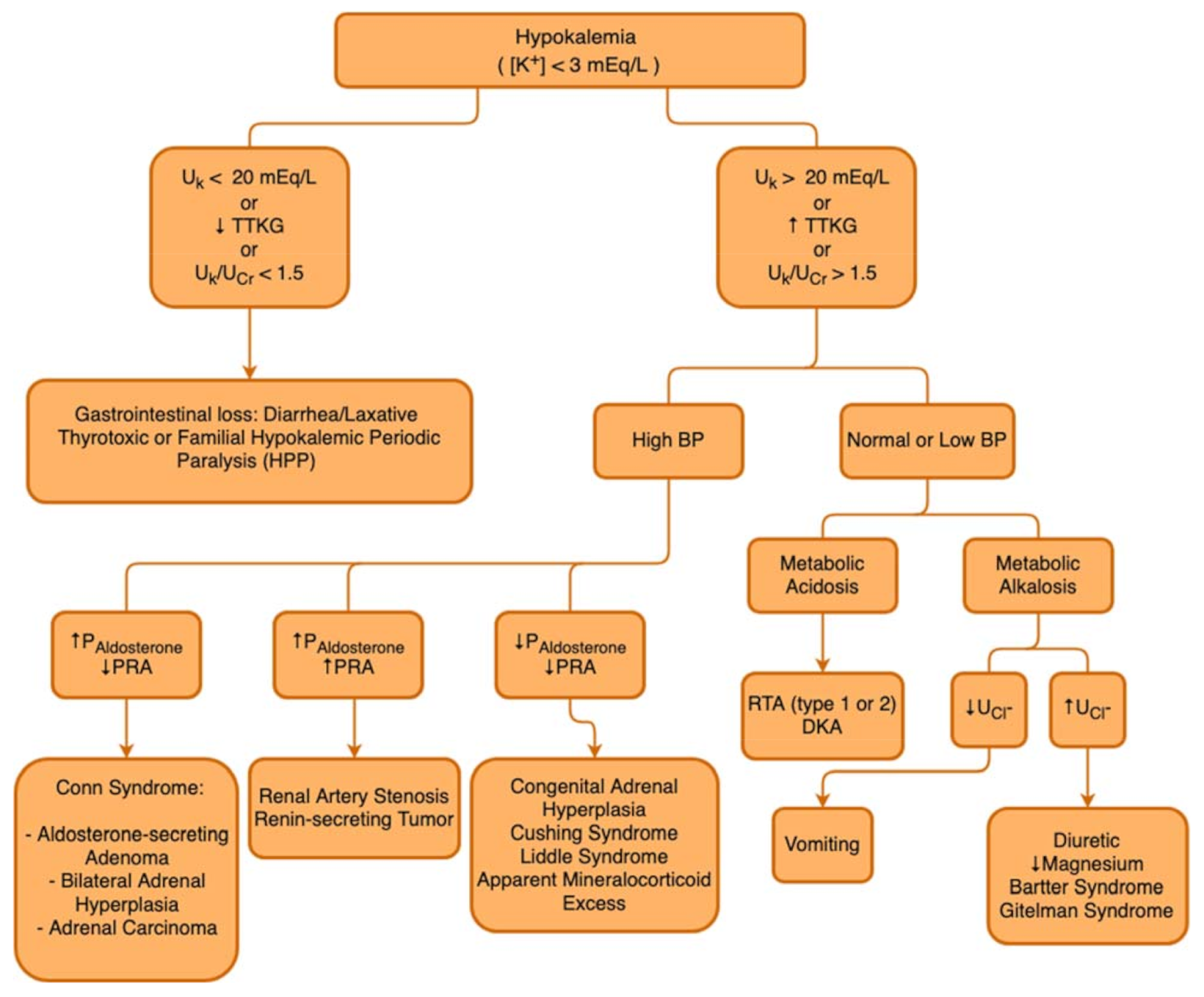
3.5. Diagnosis of Hypokalemia
3.5.1. Fractional Excretion of Potassium (FEK)
3.5.2. Transtubular Potassium Gradient (TTKG)
3.6. Management of Hypokalemia
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saeedi, P.; Salpea, P.; Karuranga, S.; Petersohn, I.; Malanda, B.; Gregg, E.W.; Unwin, N.; Wild, S.H.; Williams, R. Mortality attributable to diabetes in 20–79 years old adults, 2019 estimates: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2020, 162, 108086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.J.; Pitt, B.; Reaven, N.; Funk, S.; McGaughey, K.; Wilson, D.; Bushinsky, D.A. Association of serum potassium with all-cause mortality in patients with and without heart failure, chronic kidney disease, and/or diabetes. Am. J. Nephrol. 2017, 46, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Marrero, S.; Cainzos-Achirica, M.; Monterde, D.; Garcia-Eroles, L.; Enjuanes, C.; Yun, S.; Garay, A.; Moliner, P.; Alcoberro, L.; Corbella, X.; et al. Real-world epidemiology of potassium derangements among chronic cardiovascular, metabolic and renal conditions: A population-based analysis. Clin. Epidemiol. 2020, 12, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Gennari, F.J. Hypokalemia. N. Engl. J. Med. 1998, 339, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Colangelo, L.A.; Yeh, H.C.; Anderson, C.A.; Daviglus, M.L.; Liu, K.; Brancati, F.L. Potassium intake and risk of incident type 2 diabetes mellitus: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. Diabetologia 2012, 55, 1295–1303. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, R.; Yeh, H.-C.; Shafi, T.; Selvin, E.; Andersen, C.; Pankow, J.S.; Miller, E.; Brancati, F. Serum and Dietary Potassium and Risk of Incident Type 2 Diabetes Mellitus: The Atherosclerosis Risk in Communities (ARIC) Study. Arch. Intern. Med. 2010, 170, 1745–1751. [Google Scholar] [CrossRef]
- Zillich, A.J.; Garg, J.; Basu, S.; Bakris, G.L.; Carter, B.L. Thiazide diuretics, potassium, and the development of diabetes: A quantitative review. Hypertension 2006, 48, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Anderson, S.D.; Wen, S.; Gong, Y.; Turner, S.T.; Cooper-Dehoff, R.M.; Schwartz, G.L.; Bailey, K.; Chapman, A.; Hall, K.L.; et al. Lack of correlation between thiazide-induced hyperglycemia and hypokalemia: Subgroup analysis of results from the pharmacogenomic evaluation of antihypertensive responses (PEAR) study. Pharmacotherapy 2009, 29, 1157–1165. [Google Scholar] [CrossRef] [Green Version]
- Gloyn, A.L.; Pearson, E.; Antcliff, J.F.; Proks, P.; Bruining, G.J.; Slingerland, A.S.; Howard, N.; Srinivasan, S.; Silva, J.M.C.L.; Molnes, J.; et al. Activating Mutations in the Gene Encoding the ATP-Sensitive Potassium-Channel Subunit Kir6.2 and Permanent Neonatal Diabetes. N. Engl. J. Med. 2014, 350, 1838–1849. [Google Scholar] [CrossRef] [Green Version]
- Weiner, I.D.; Wingo, C.S. Hypokalemia-Consequences Causes, and correction. J. Am. Soc. Nephrol. 1997, 8, 1179–1188. [Google Scholar] [CrossRef]
- Viera, A.J.; Wouk, N. Potassium disorders: Hypokalemia and hyperkalemia. Am. Fam. Physician 2015, 92, 487–495. [Google Scholar] [PubMed]
- Eslam, R.B.; Öztürk, B.; Panzer, S.; Qin, H.; Duca, F.; Binder, C.; Rettl, R.; Dachs, T.M.; Alasti, F.; Vila, G.; et al. Low serum potassium levels and diabetes—An unfavorable combination in patients with heart failure and preserved ejection fraction. Int. J. Cardiol. 2020, 317, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Muneer, M.; Akbar, I. Acute Metabolic Emergencies in Diabetes: DKA, HHS and EDKA. Exp. Med. Biol. 2021, 1307, 85–114. [Google Scholar]
- Ellison, D.H. Clinical pharmachology in diuretic use. Clin. J. Am. Soc. Nephrol. 2019, 14, 1248–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, B.F. Metabolic complications associated with use of diuretics. Semin. Nephrol. 2011, 31, 542–552. [Google Scholar] [CrossRef]
- Barzilay, J.I.; Davis, B.R.; Pressel, S.L.; Cutler, J.A.; Einhorn, P.T.; Black, H.R.; Cushman, W.C.; Ford, C.E.; Margolis, K.L.; Moloo, J.; et al. ALLHAT Collaborative Research Group. Long-term effects of incident diabetes mellitus on cardiovascular outcomes in people treated for hypertension: The ALLHAT Diabetes Extension Study. Cardiovasc. Qual. Outcomes 2012, 5, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Hripcsak, G.; Suchard, M.A.; Shea, S.; Chen, R.; You, S.C.; Pratt, N.; Madigan, D.; Krumholz, H.M.; Ryan, P.B.; Schuemie, M.J. Comparison of Cardiovascular and Safety Outcomes of Chlorthalidone vs Hydrochlorothiazide to Treat Hypertension. JAMA Intern. Med. 2020, 180, 542–551. [Google Scholar] [CrossRef]
- De Lucena, D.D.; Rangel, É.B. Glucocorticoids use in kidney transplant setting. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1023–1041. [Google Scholar] [CrossRef]
- Nazari, S.; Rameshrad, M.; Hosseinzadeh, H. Toxicological Effects of Glycyrrhiza glabra (Licorice): A Review. Phytother. Res. 2017, 31, 1635–1650. [Google Scholar] [CrossRef]
- Kim, Y.W. Antimicrobial-induced Electrolyte and Acid-Base Disturbances. Electrolyte Blood Press. 2008, 5, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Kerstan, D.; Dai, L.; Ritchie, G.; Quamme, G.A. Aminoglycosides inhibit hormone-stimulated Mg2+ uptake in mouse distal convoluted tubule cells. Can. J. Physiol. Pharmacol. 2000, 78, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Muskiet, M.H.; Tonneijck, L.; Smits, M.M.; Kramer, M.H.; Ouwens, D.M.; Hartmann, B.; Holst, J.J.; Touw, D.J.; Danser, A.J.; Joles, J.A.; et al. Effects of DPP-4 Inhibitor Linagliptin Versus Sulfonylurea Glimepiride as Add-on to Metformin on Renal Physiology in Overweight Patients with Type 2 Diabetes (RENALIS): A Randomized, Double-Blind Trial. Diabetes Care 2020, 43, 2889–2893. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Ghaleb, R.; Mansour, H.; Hanafy, A.; Mahmoud, N.M.; Elsharef, M.A.; Salama, M.K.; Elsaughier, S.M.; Abdel-Wahid, L.; Mohamed, M.E.; et al. Safety and Efficacy of Adding Dapagliflozin to Furosemide in Type 2 Diabetic Patients with Decompensated Heart Failure and Reduced Ejection Fraction. Front. Cardiovasc. Med. 2020, 7, 602251. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.; Rao, V.S.; Ivey-Miranda, J.; Fleming, J.; Mahoney, D.; Maulion, C.; Suda, N.; Siwakoti, K.; Ahmad, T.; Jacoby, D.; et al. Empagliflozin in Heart Failure: Diuretic and Cardiorenal Effects. Circulation 2020, 142, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Cox, Z.L.; Collins, S.P.; Aaron, M.; Hernandez, G.A.; McRae, A.T., III; Davidson, B.T.; Fowler, M.; Lindsell, C.J.; Harrell, F.E., Jr.; Jenkins, C.A.; et al. Efficacy and safety of dapagliflozin in acute heart failure: Rationale and design of the DICTATE-AHF trial. Am. Heart J. 2021, 232, 116–124. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, J.; Liu, W.; Su, X. Determining the Prevalence of Primary Aldosteronism in Patients with New-Onset Type 2 Diabetes and Hypertension. J. Clin. Endocrinol. Metab. 2020, 105, dgz293. [Google Scholar] [CrossRef]
- Akehi, Y.; Yanase, T.; Motonaga, R.; Umakoshi, H.; Tsuiki, M.; Takeda, Y.; Yoneda, T.; Kurihara, I.; Itoh, H.; Katabami, T.; et al. TJapan Primary Aldosteronism Study Group. High Prevalence of Diabetes in Patients with Primary Aldosteronism (PA) Associated With Subclinical Hypercortisolism and Prediabetes More Prevalent in Bilateral Than Unilateral PA: A Large, Multicenter Cohort Study in Japan. Diabetes Care 2019, 42, 938–945. [Google Scholar]
- Sebastian, A.; McSherry, E.; Morris, R.C.J. Renal potassium wasting in renal tubular acidosis (RTA): Its occurrence in types 1 and 2 RTA despite sustained correction of systemic acidosis. J. Clin. Investig. 1971, 50, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Kobrin, S.M.; Goldfarb, S. Magnesium deficiency. Semin. Nephrol. 1990, 10, 525–535. [Google Scholar]
- Shardha, A.K.; Vaswani, A.S.; Faraz, A.; Alam, M.T.; Kumar, P. Frequency and risk factors associated with hypomagnesaemia in hypokalemic type-2 diabetic patients. J. Coll. Physicians Surg. Pak. 2014, 24, 830–835. [Google Scholar]
- González, W.; Altieri, P.I.; Alvarado, S.; Banchs, H.L.; Escobales, N.; Crespo, M.; Borges, W. Magnesium: The forgotten electrolyte. Bol. Asoc. Med. Puerto Rico 2013, 105, 17–20. [Google Scholar]
- Huang, C.-L.; Kio, E. Mechanism of hypokalemia in magnesium deficiency. J. Am. Soc. Nephrol. 2007, 18, 2649–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-H.; Yang, S.-S.; Chau, T. A practical approach to genetic hypokalemia. Electrolyte Blood Press 2010, 8, 38–50. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Gang, X.; Sun, Z.; Wang, P.; Wang, G.; Guo, W. Type 2 diabetes mellitus caused by Gitelman syndrome-related hypokalemia A case report. Medicine 2020, 99, e21123. [Google Scholar] [CrossRef]
- Hitomi, H.; Kiyomoto, H.; Nishiyama, A.; Hara, T.; Moriwaki, K.; Kaifu, K.; Ihara, G.; Fujita, Y.; Ugawa, T.; Kohno, M. Aldosterone suppresses insulin signaling via the downregulation of insulin receptor substrate-1 in vascular smooth muscle cells. Hypertension 2007, 50, 750–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanesi, A.; Weinreb, J.E. Hyperglycemic Hyperosmolar State. In Comprehensive FREE Online Endocrinology Book; Feingold, K.R.F., Anawalt, B., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2018. [Google Scholar]
- Jang, T.B.; Chauhan, V.; Morchi, R.; Najand, H.; Naunheim, R.; Kaji, A.H. Hypokalemia in diabetic ketoacidosis is less common than previously reported. Intern. Emerg. Med. 2015, 10, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Marti, G.; Schwarz, C.; Leichtle, A.B.; Fiedler, G.-M.; Arampatzis, S.; Exadaktylos, A.K.; Lindner, G. Etiology and symptoms of severe hypokalemia in emergency department patients. Eur. J. Emerg. Med. 2014, 21, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Al-Azzawi, O.F.N.; Razak, M.K.A.; Al-Hammady, S.J. Rhabdomyolysis; is it an overlooked DKA complication. Diabetes Metab. Syndr. 2019, 13, 3047–3052. [Google Scholar] [CrossRef]
- Lim, S. Approach to hypokalemia. Acta Med. Indones 2007, 39, 56–64. [Google Scholar]
- Kardalas, E.; Paschou, S.A.; Anagnostis, P.; Muscogiuri, G.; Siasos, G.; Vryonidou, A. Hypokalemia: A clinical update. Endocr. Connect. 2018, 7, R135–R146. [Google Scholar] [CrossRef]
- Liamis, G.; Liberopoulos, E.; Barkas, F.; Elisaf, M. Diabetes mellitus and electrolyte disorders. World J. Clin. Cases 2014, 2, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Xu, X.; Wang, X. Altered mRNA Expression of ATP-Sensitive and Inward Rectifier Potassium Channel Subunits in Streptozotocin-Induced Diabetic Rat Heart and Aorta. J. Pharmacol. Sci. 2003, 93, 478–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unwin, R.J.; Luft, F.C.; Shirley, D.G. Pathophysiology and management of hypokalemia: A clinical perspective. Nat. Rev. Nephrol. 2011, 7, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Assadi, F. Diagnosis of hypokalemia: A problem-solving approach to clinical cases. Iran. J. Kidney Dis. 2008, 2, 115–122. [Google Scholar] [PubMed]
- Usman, A. Initial potassium replacement in diabetic ketoacidosis: The unnoticed area of gap. Front. Endocrinol. 2018, 9, 109. [Google Scholar] [CrossRef]
- National Collaborating Centre for Women’s and Children’s Health (UK). Type 1 Diabetes: Diagnosis and Management of Type 1 Diabetes in Children and Young People; RCOG Press: London, UK, 2004. [Google Scholar]
- Kitabchi, A.E.; Umpierrez, G.E.; Murphy, M.B.; Kreisberg, R.A. Hyperglycemic crises in adult patients with diabetes: A consensus statement from the American Diabetes Association. Diabetes Care 2006, 29, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Savage, M.W.; Dhatariya, K.K.; Kilvert, A.; Rayman, G.; Rees, J.A.; Courtney, C.H.; Hilton, L.; Dyer, P.H.; Hamersley, M.S. Joint British Diabetes Societies guideline for the management of diabetic ketoacidosis. Diabet. Med. 2011, 28, 508–515. [Google Scholar] [CrossRef]
- Malaysia MoH. Clinical Practice Guideline Management of Type 2 Diabetes Mellitus, 5th ed.; MOH/P/PAK/184.09 (GU). 2015. Available online: https://www.moh.gov.my/moh/resources/Penerbitan/CPG/Endocrine/3a.pdf (accessed on 13 October 2021).


{kind=link}
{kind=link}
{kind=link}
| Drug-Induced Transcellular Shifts | Induced Gastrointestinal Losses |
|---|---|
| Insulin Beta-2 agonists Verapamil Chloroquine Barium | Laxatives Enemas Prolonged vomiting Volume depletion |
| Nondrug-Induced Transcellular Shifts | Drug-Induced Potassium Losses |
| Neoplasms Thyrotoxicosis Primary hyperaldosteronism Familial hypokalemic paralysis Delirium tremens Barium intoxication Cushing syndrome | Thiazide diuretics Loop diuretics Glucocorticoids Mineralocorticoids Penicillin Aminoglycosides Amphotericin B Glycyrhizza glabra |
| Nondrug-Induced Potassium Losses | |
| Low dietary intake Diarrhea Metabolic alkalosis Type I and II renal tubular acidosis Hypomagnesemia Acute and chronic complications of DM Exocrine pancreatic insufficiency Infections Inflammatory bowel diseases Malabsorptive syndromes Bartter syndrome Gitelman syndrome Acute tubular injuries |
| Diagnostic Criteria | HHS | DKA |
|---|---|---|
| pH | >7.30 | ≤7.30 |
| Plasma Glucose | >540–600 mg/dL | >250 mg/dL |
| Serum Bicarbonate | >15–18 mEq/L | <18 mEq/L |
| Plasma and Urine Ketones | None or trace | Positive |
| Anion Gap: Na+ − (Cl− + HCO3−) | <12 | >12 |
| Serum osmolality | >320 mOsm/Kg | Variable |
| Glycosuria | ++ | ++ |
| Typical Deficit | ||
| Water (mL/Kg) | 100–200 (9 L) | 100 (6 L) |
| Na+ (mEq/Kg) | 5–13 | 7–10 |
| Cl− (mEq/Kg) | 5–15 | 3–5 |
| K+ (mEq/Kg) | 4–6 | 3–5 |
| PO4− (mmol/Kg) | 3–7 | 5–7 |
| Mg2+ and Ca2+ | 1–2 | 1–2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coregliano-Ring, L.; Goia-Nishide, K.; Rangel, É.B. Hypokalemia in Diabetes Mellitus Setting. Medicina 2022, 58, 431. https://doi.org/10.3390/medicina58030431
Coregliano-Ring L, Goia-Nishide K, Rangel ÉB. Hypokalemia in Diabetes Mellitus Setting. Medicina. 2022; 58(3):431. https://doi.org/10.3390/medicina58030431
Chicago/Turabian StyleCoregliano-Ring, Lucas, Kleber Goia-Nishide, and Érika Bevilaqua Rangel. 2022. "Hypokalemia in Diabetes Mellitus Setting" Medicina 58, no. 3: 431. https://doi.org/10.3390/medicina58030431
APA StyleCoregliano-Ring, L., Goia-Nishide, K., & Rangel, É. B. (2022). Hypokalemia in Diabetes Mellitus Setting. Medicina, 58(3), 431. https://doi.org/10.3390/medicina58030431