
Screening of Relevant Metabolism-Disrupting Chemicals on Pancreatic β-Cells: Evaluation of Murine and Human In Vitro Models

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
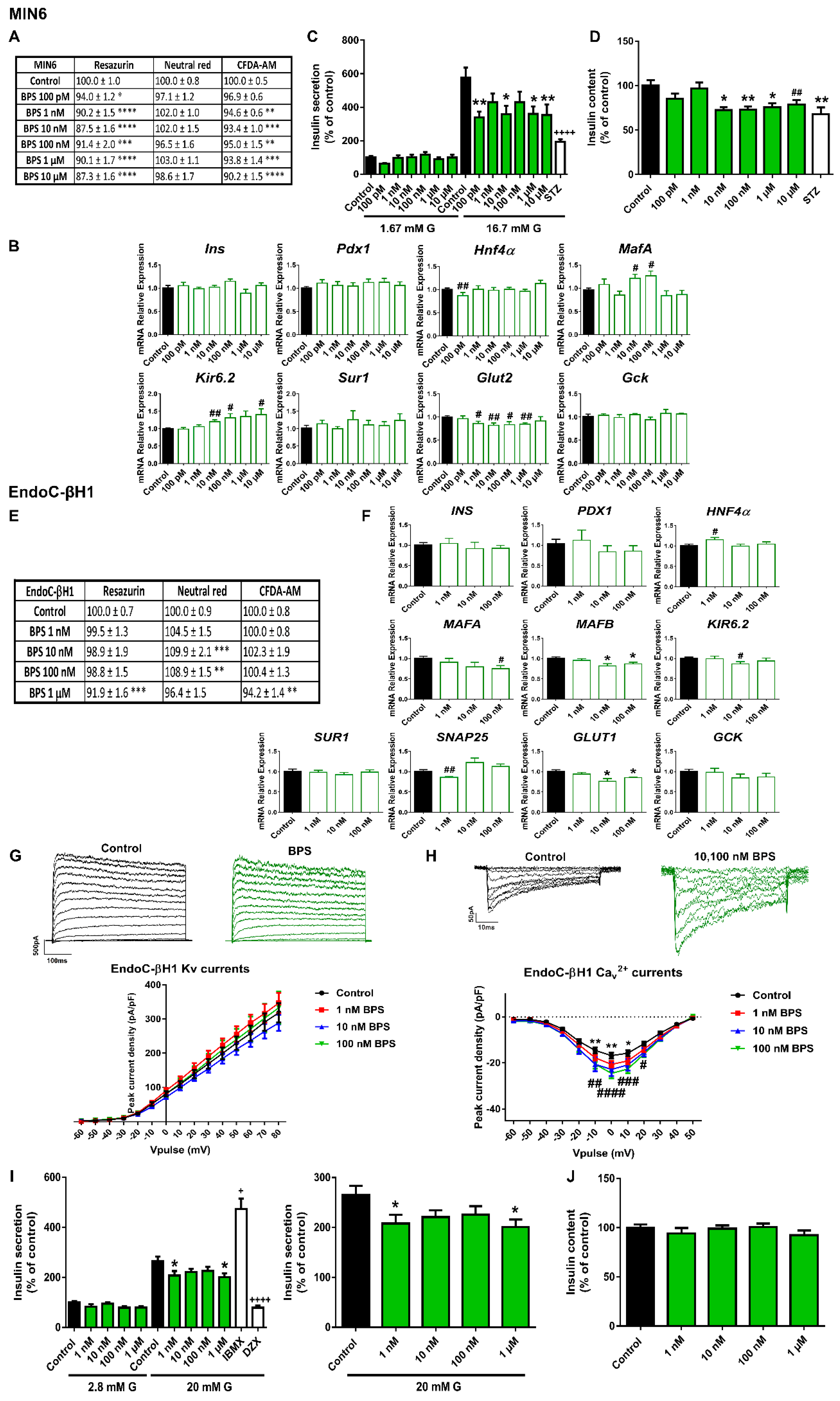
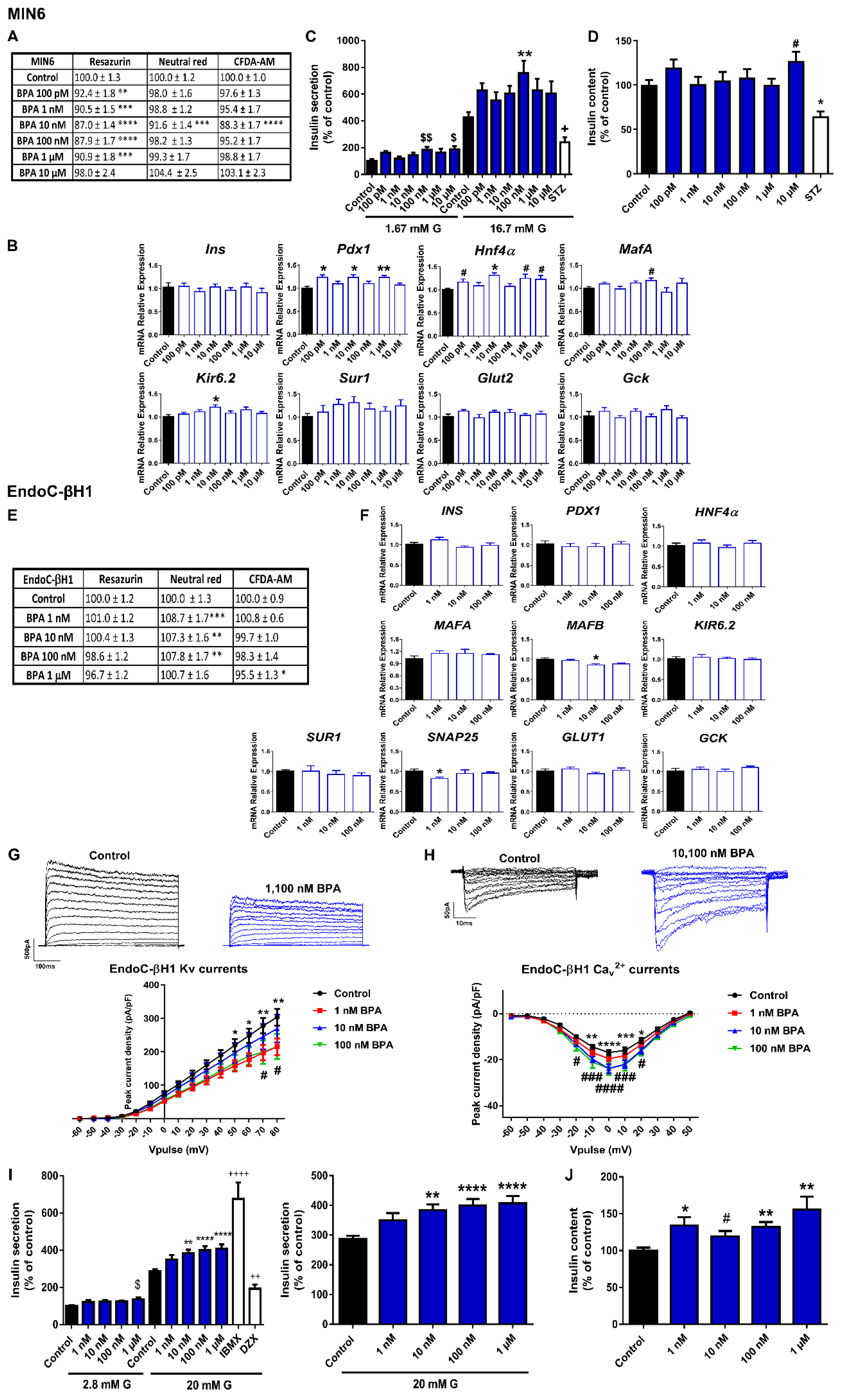
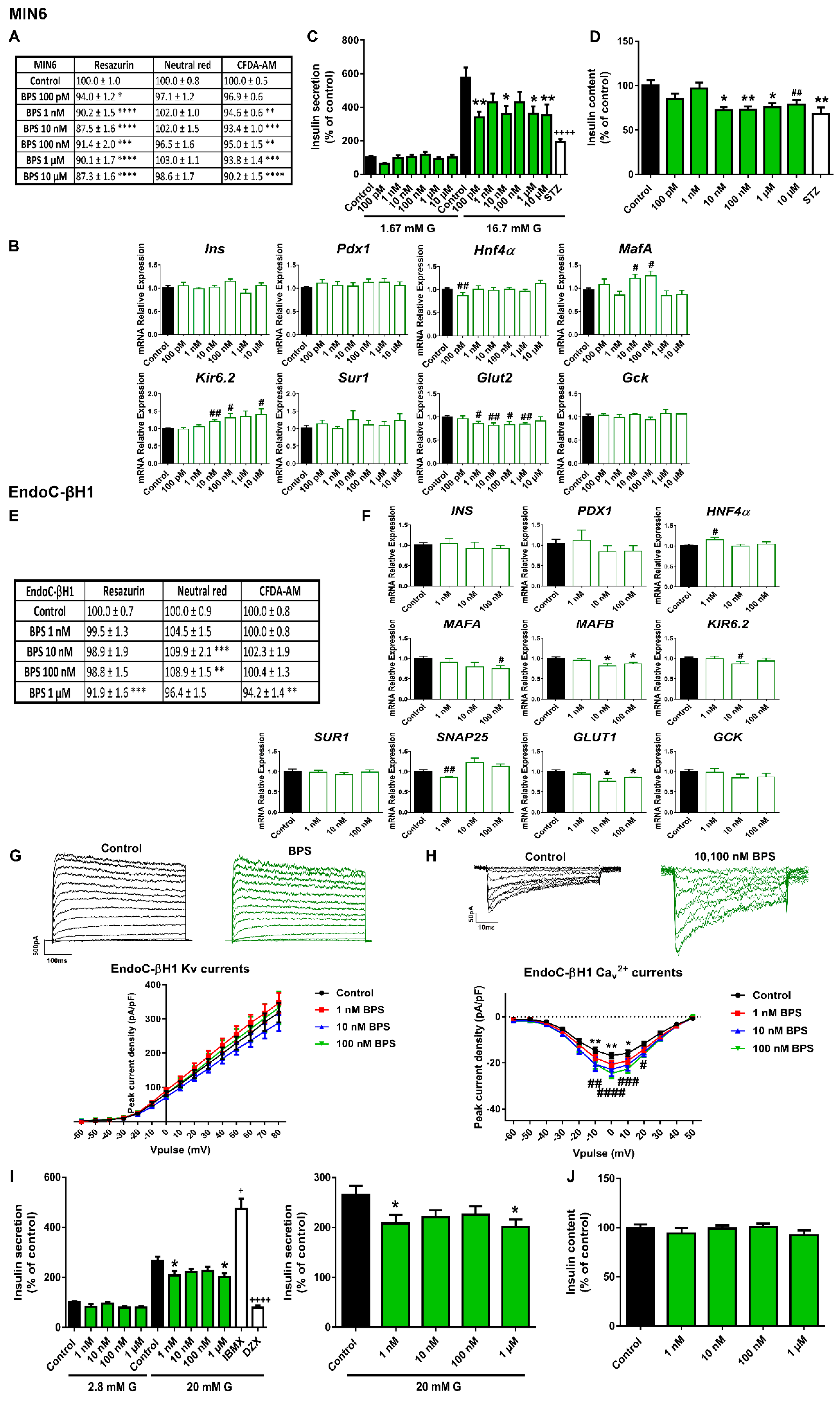
2.1. Bisphenols A and S Alter Pancreatic β-Cell Function in Both Mouse and Human Pancreatic β-Cells
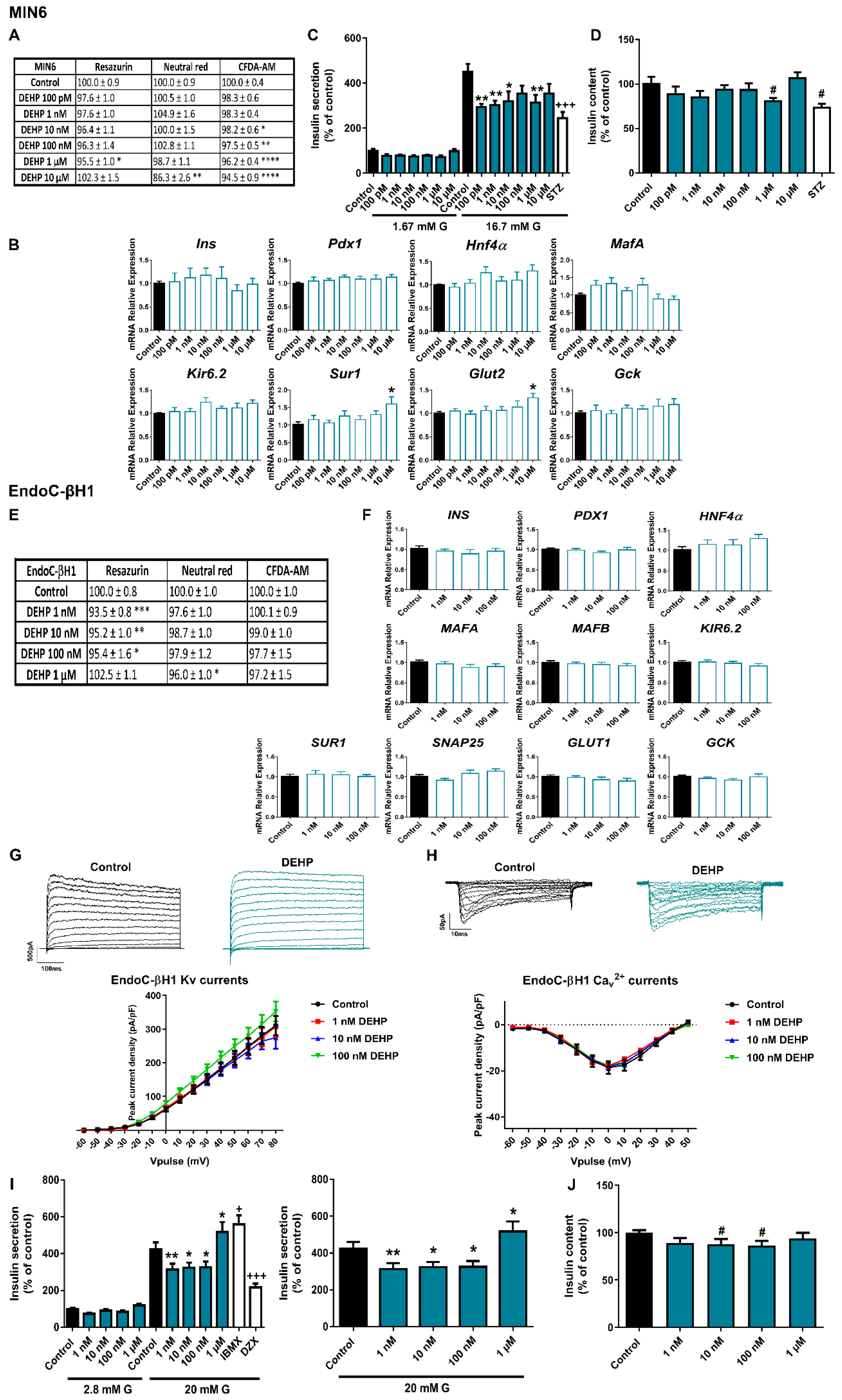
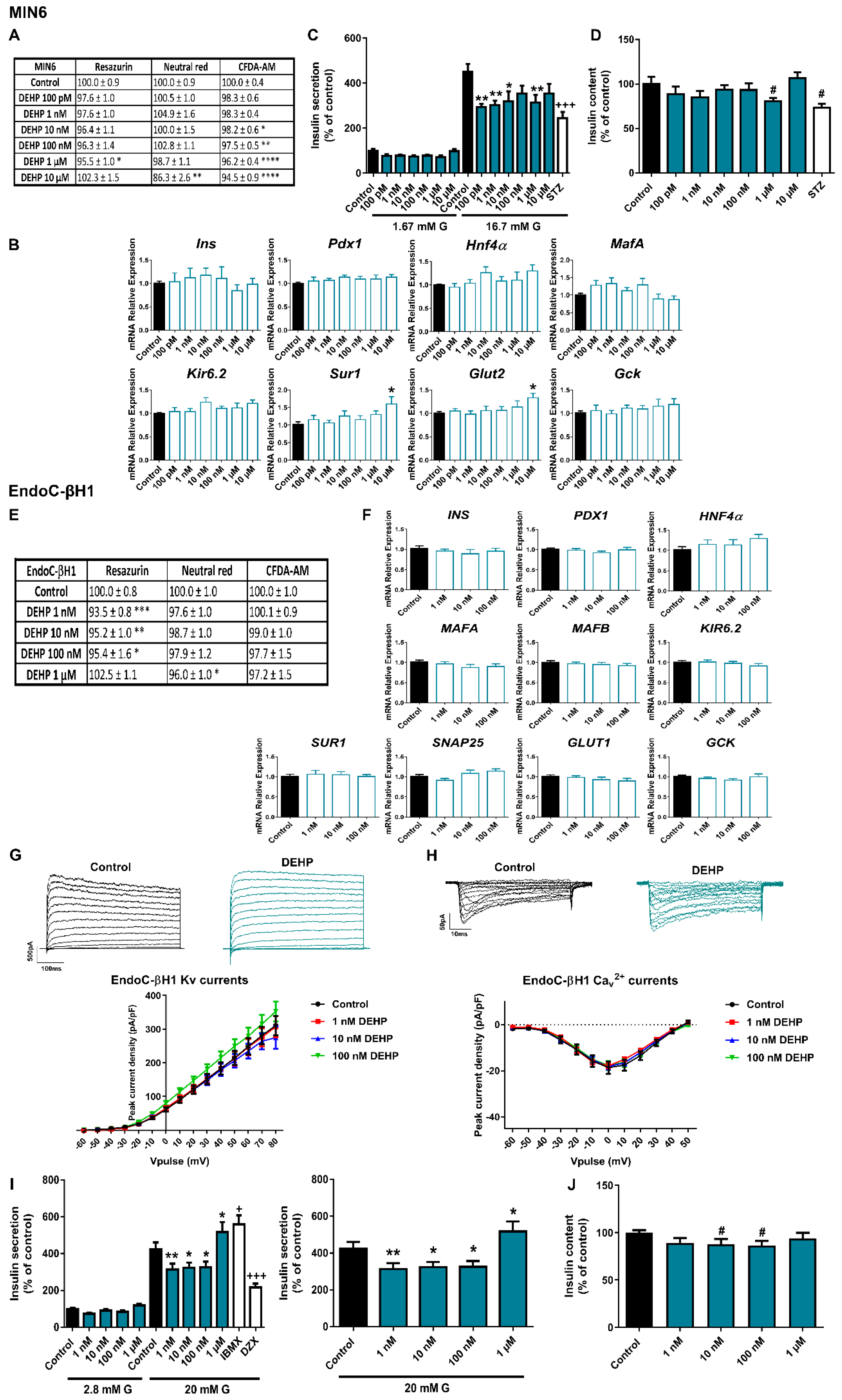
2.2. The Phthalate DEHP Disrupts Murine and Human Pancreatic β-Cell Function in a Similar Manner
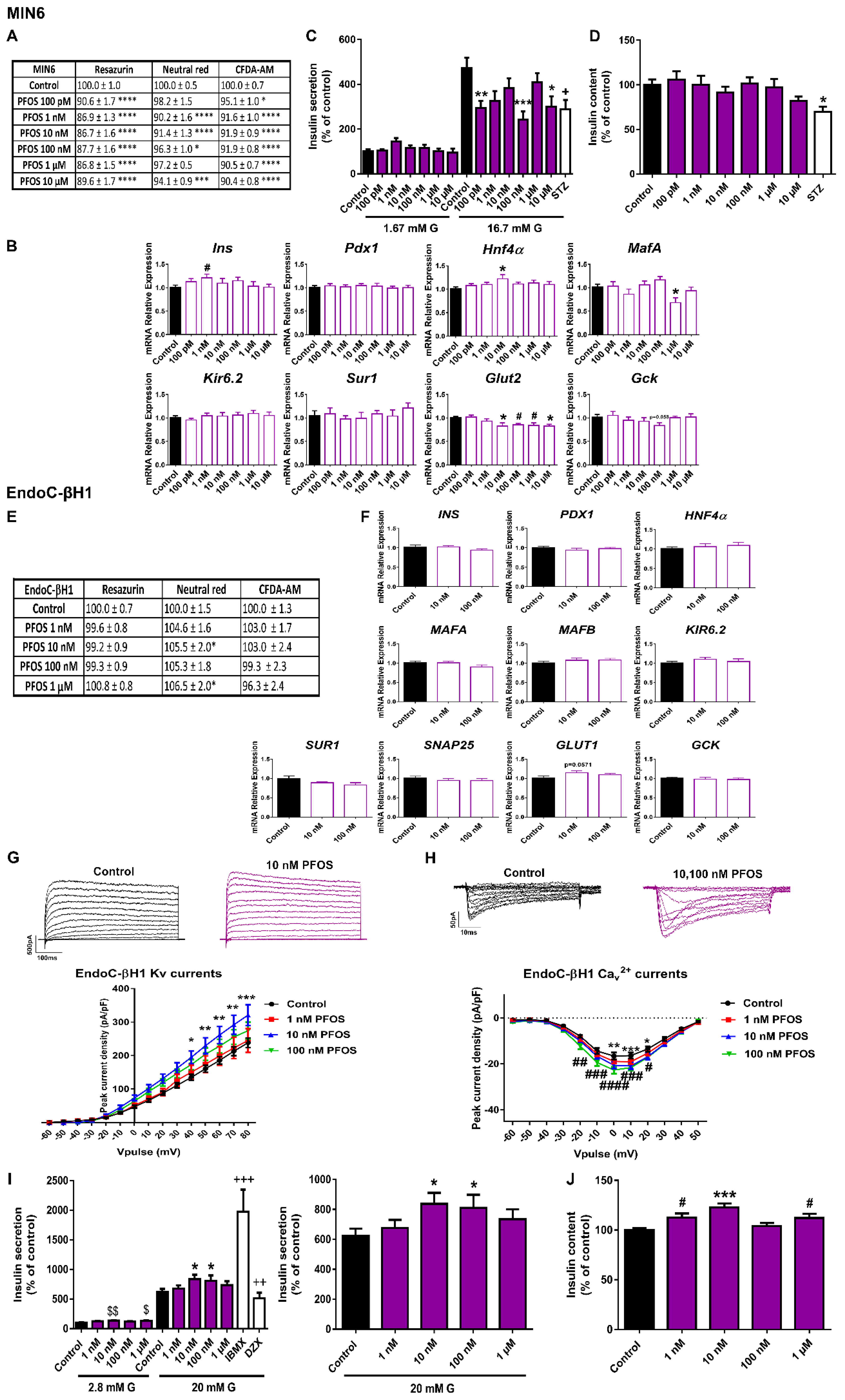
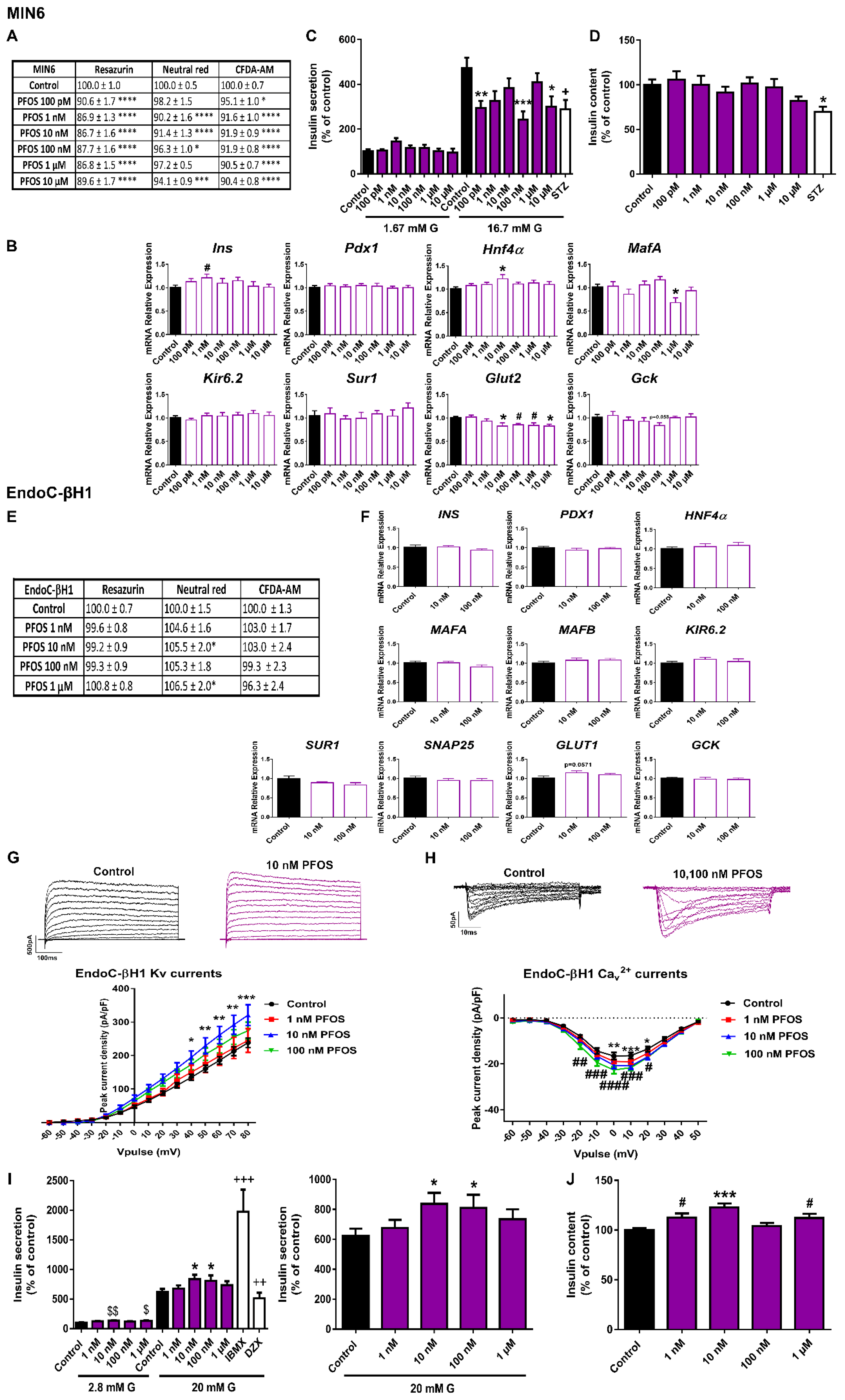
2.3. Different Effects of PFOS Exposure on Human and Murine Pancreatic β-Cell Models
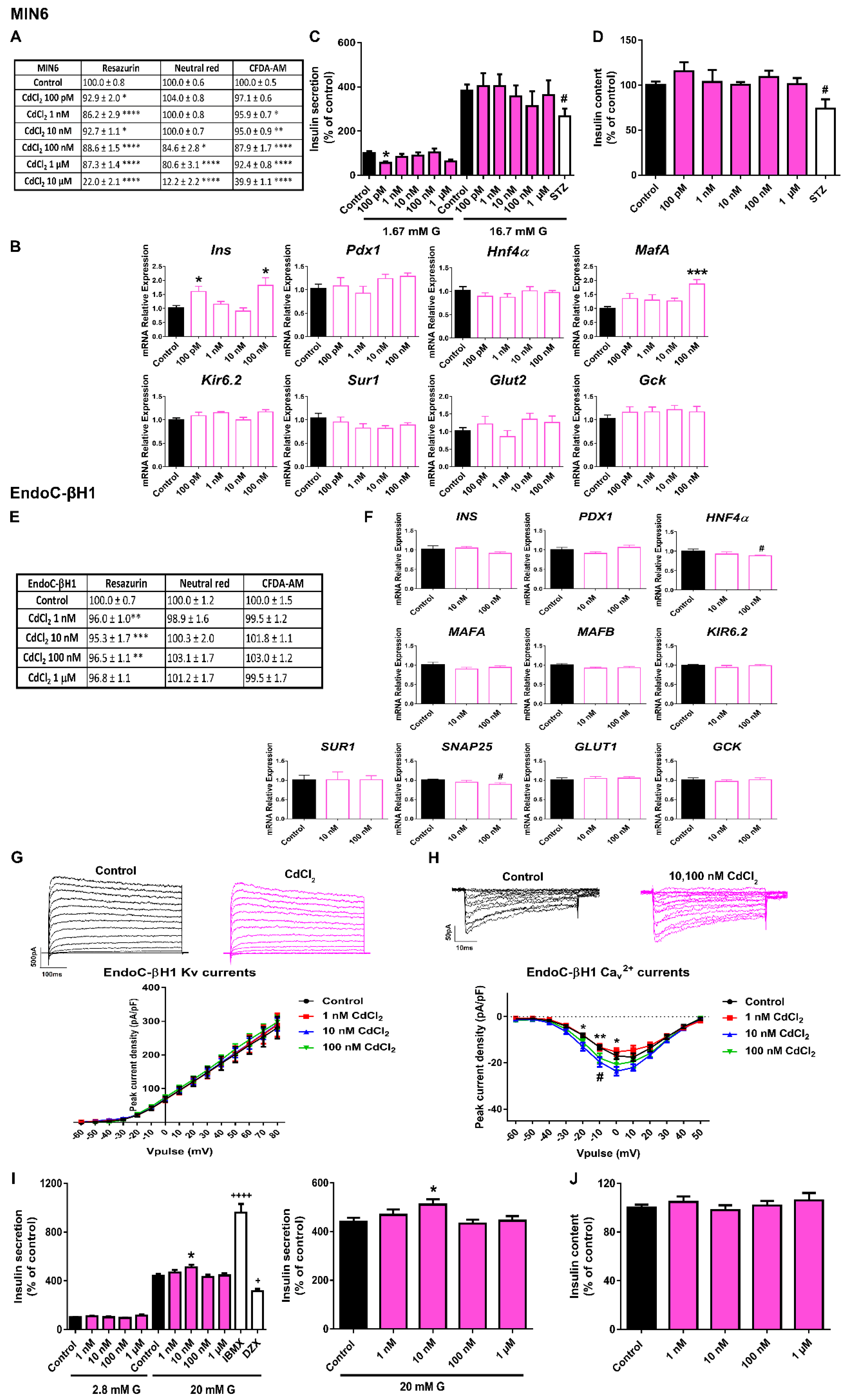
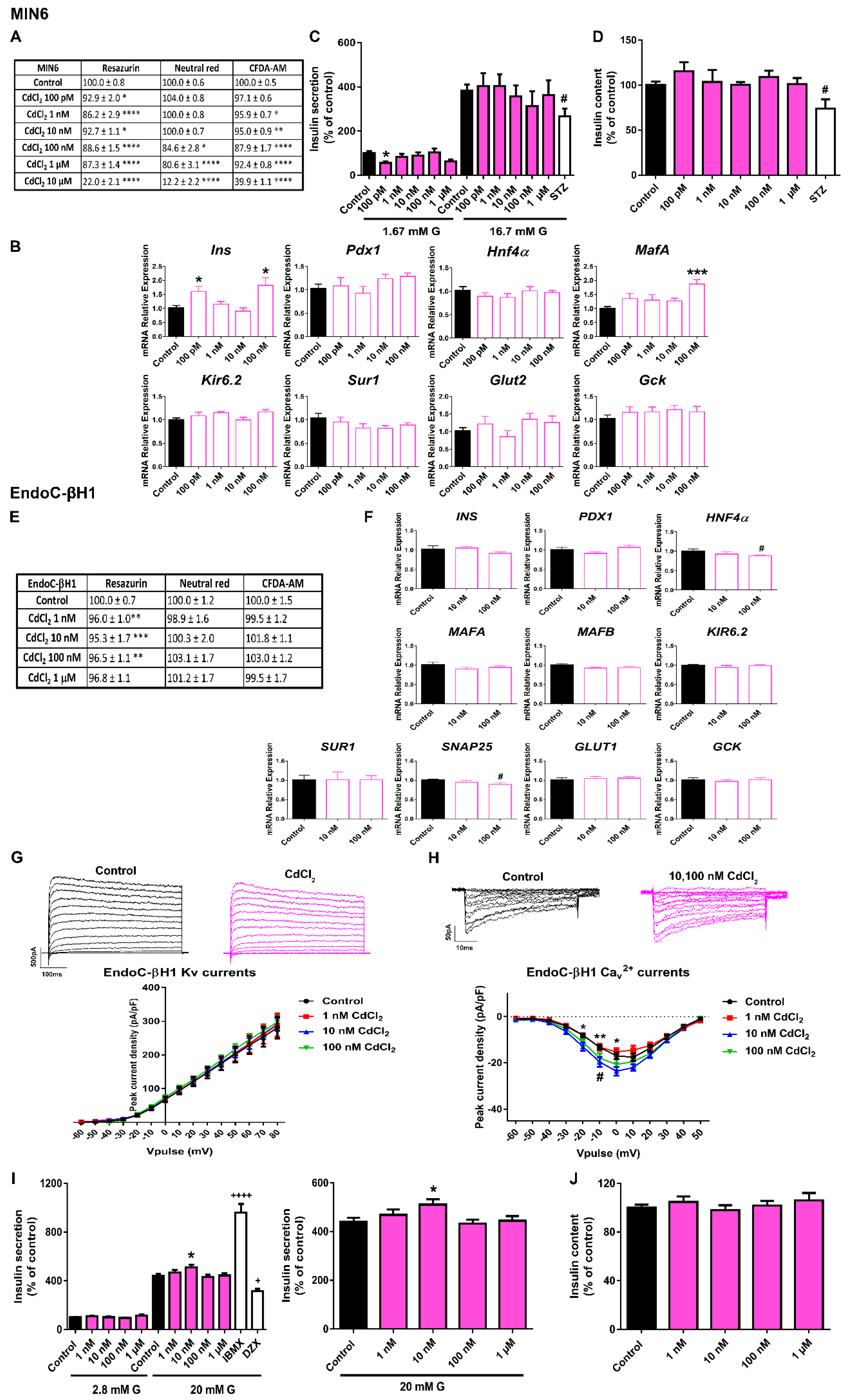
2.4. CdCl2 Effects on Pancreatic β-Cell Function and Viability
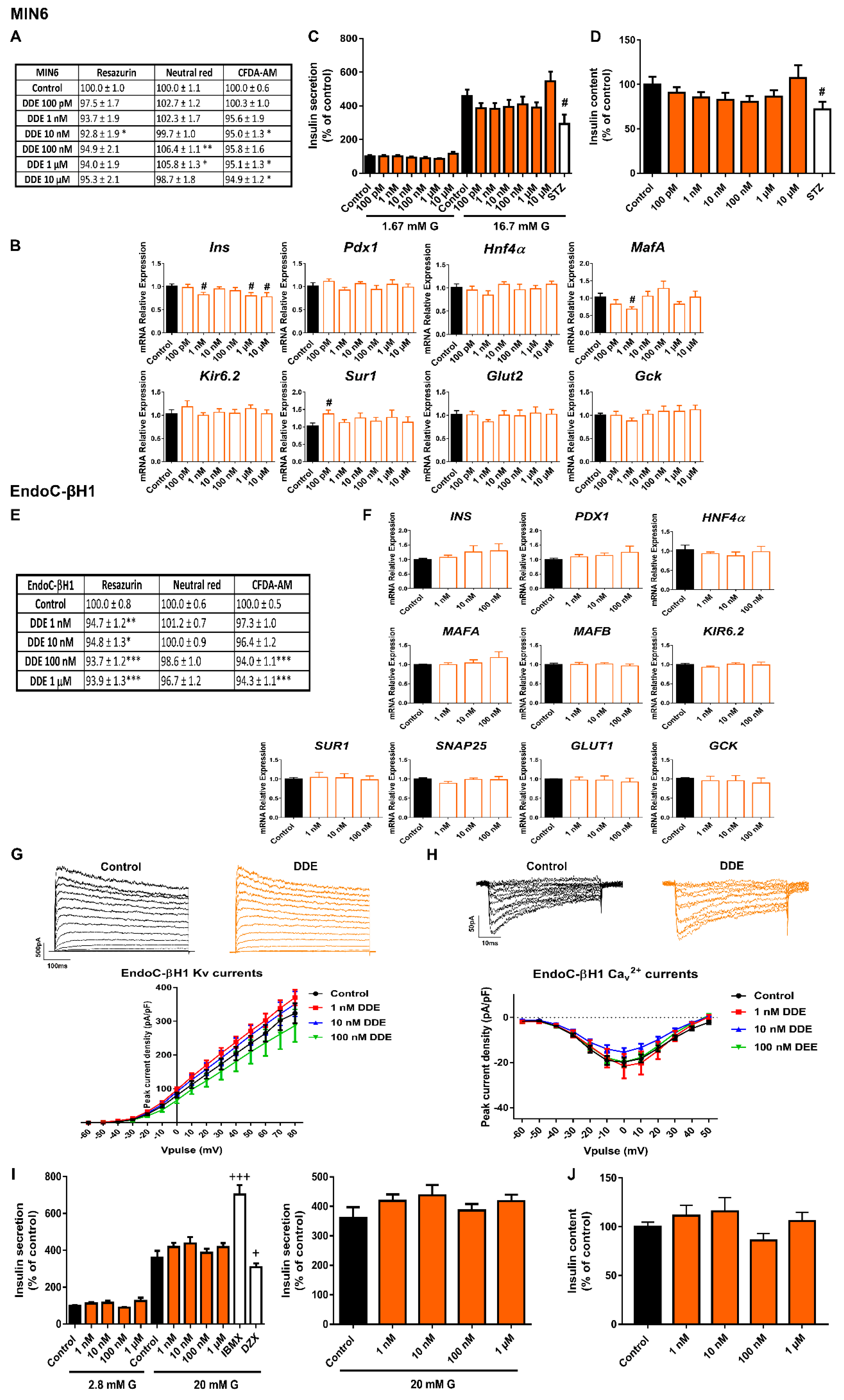
2.5. DDE Exposure Did Not Compromise Pancreatic β-Cell Function or Viability
3. Discussion
3.1. Bisphenols:
3.2. DEHP
3.3. PFOS
3.4. CdCl2
3.5. DDE
4. Conclusions
5. Material and Methods
5.1. Chemicals
5.2. Cell Culture
5.3. Cell Viability
5.4. Insulin Secretion and Content
5.5. RNA Extraction and RT-qPCR
5.6. Electrophysiological Recordings: K+ and Ca2+ Currents
5.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AO | Adverse outcome |
| AOP | Adverse outcome pathway |
| BPA | Bisphenol-A |
| BPF | Bisphenol-F |
| BPS | Bisphenol-S |
| CdCl2 | Cadmium chloride |
| CFDA-AM | Carboxyfluorescein diacetate acetoxymethyl ester |
| DDE | Dichlorodiphenyldichloroethylene |
| DEHP | Di(2-ethylhexyl) phthalate |
| EDCs | Endocrine-disrupting chemicals |
| EndoC-βH1 | Human pancreatic β cell line EndoC-βH1 |
| GSIS | Glucose-stimulated insulin secretion |
| KEs | Key events |
| MDCs | Metabolism-disrupting chemicals |
| MIN6 | Mouse insulinoma 6 |
| NRU | Neutral red uptake |
| OECD | Organization for Economic Cooperation and Development |
| PFOS | Perfluorooctanesulfonic acid |
| RZ | Resazurin |
References
- MacDonald, P.E.; Joseph, J.W.; Rorsman, P. Glucose-sensing mechanisms in pancreatic beta-cells. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2005, 360, 2211–2225. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rorsman, P. Molecular defects in insulin secretion in type-2 diabetes. Rev. Endocr. Metab. Disord. 2004, 5, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E. The importance of the beta-cell in the pathogenesis of type 2 diabetes mellitus. Am. J. Med. 2000, 108 (Suppl. S6a), 2S–8S. [Google Scholar] [CrossRef]
- Prentki, M.; Peyot, M.L.; Masiello, P.; Madiraju, S.R.M. Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic beta-Cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef]
- Hudish, L.I.; Reusch, J.E.; Sussel, L. Beta Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J. Clin. Investig. 2019, 129, 4001–4008. [Google Scholar] [CrossRef] [Green Version]
- OECD/EuropeanUnion. Health at a Glance: Europe 2020: State of Health in the EU Cycle; OECD Publishing: Paris, France, 2020. [Google Scholar] [CrossRef]
- CDC. National Diabetes Statistics Report. 2020. Available online: https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf (accessed on 13 February 2022).
- WHO, World Health Organization. Obesity and Overweight. 2016. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 13 February 2022).
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Franks, P.W.; McCarthy, M.I. Exposing the exposures responsible for type 2 diabetes and obesity. Science 2016, 354, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, A. Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 7, 346–353. [Google Scholar] [CrossRef]
- Nadal, A.; Quesada, I.; Tuduri, E.; Nogueiras, R.; Alonso-Magdalena, P. Endocrine-disrupting chemicals and the regulation of energy balance. Nat. Rev. Endocrinol. 2017, 13, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- OECD. Revised Guidance Document 150 on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption. 2017. Available online: https://www.oecd-ilibrary.org/environment/revised-guidance-document-150-on-standardised-test-guidelines-for-evaluating-chemicals-for-endocrine-disruption/introduction_9789264304741-1-en (accessed on 13 February 2022).
- Green, A.D.; Vasu, S.; Flatt, P.R. Cellular models for beta-cell function and diabetes gene therapy. Acta Physiol. 2018, 222, e13012. [Google Scholar] [CrossRef]
- Rorsman, P.; Ashcroft, F.M. Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- Yang, S.N.; Shi, Y.; Yang, G.; Li, Y.; Yu, J.; Berggren, P.O. Ionic mechanisms in pancreatic beta cell signaling. Cell. Mol. Life Sci. CMLS 2014, 71, 4149–4177. [Google Scholar] [CrossRef]
- Scharfmann, R.; Staels, W.; Albagli, O. The supply chain of human pancreatic beta cell lines. J. Clin. Investig. 2019, 129, 3511–3520. [Google Scholar] [CrossRef] [Green Version]
- Tsonkova, V.G.; Sand, F.W.; Wolf, X.A.; Grunnet, L.G.; Ringgaard, A.K.; Ingvorsen, C.; Winkel, L.; Kalisz, M.; Dalgaard, K.; Bruun, C.; et al. The EndoC-betaH1 cell line is a valid model of human beta cells and applicable for screenings to identify novel drug target candidates. Mol. Metab. 2018, 8, 144–157. [Google Scholar] [CrossRef]
- Hastoy, B.; Godazgar, M.; Clark, A.; Nylander, V.; Spiliotis, I.; van de Bunt, M.; Chibalina, M.V.; Barrett, A.; Burrows, C.; Tarasov, A.I.; et al. Electrophysiological properties of human beta-cell lines EndoC-betaH1 and -betaH2 conform with human beta-cells. Sci. Rep. 2018, 8, 16994. [Google Scholar] [CrossRef]
- Martinez-Pinna, J.; Marroqui, L.; Hmadcha, A.; Lopez-Beas, J.; Soriano, S.; Villar-Pazos, S.; Alonso-Magdalena, P.; Dos Santos, R.S.; Quesada, I.; Martin, F.; et al. Oestrogen receptor beta mediates the actions of bisphenol-A on ion channel expression in mouse pancreatic beta cells. Diabetologia 2019, 62, 1667–1680. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.; Ramracheya, R.; Bengtsson, M.; Zhang, Q.; Karanauskaite, J.; Partridge, C.; Johnson, P.R.; Rorsman, P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 2008, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, P.E.; Wheeler, M.B. Voltage-dependent K(+) channels in pancreatic beta cells: Role, regulation and potential as therapeutic targets. Diabetologia 2003, 46, 1046–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammala, C.; Eliasson, L.; Bokvist, K.; Larsson, O.; Ashcroft, F.M.; Rorsman, P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B-cells. J. Physiol. 1993, 472, 665–688. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, E.; Soriano, S.; Almagro, L.; Garcia-Heredia, A.; Rafacho, A.; Alonso-Magdalena, P.; Nadal, A.; Quesada, I. The effects of aging on male mouse pancreatic beta-cell function involve multiple events in the regulation of secretion: Influence of insulin sensitivity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2021, 77, 405–415. [Google Scholar] [CrossRef]
- Kolb, H.; Kempf, K.; Rohling, M.; Martin, S. Insulin: Too much of a good thing is bad. BMC Med. 2020, 18, 224. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Morimoto, S.; Ripoll, C.; Fuentes, E.; Nadal, A. The estrogenic effect of bisphenol A disrupts pancreatic beta-cell function in vivo and induces insulin resistance. Environ. Health Perspect. 2006, 114, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Magdalena, P.; Ropero, A.B.; Carrera, M.P.; Cederroth, C.R.; Baquie, M.; Gauthier, B.R.; Nef, S.; Stefani, E.; Nadal, A. Pancreatic insulin content regulation by the estrogen receptor ER alpha. PLoS ONE 2008, 3, e2069. [Google Scholar] [CrossRef] [Green Version]
- Villar-Pazos, S.; Martinez-Pinna, J.; Castellano-Munoz, M.; Alonso-Magdalena, P.; Marroqui, L.; Quesada, I.; Gustafsson, J.A.; Nadal, A. Molecular mechanisms involved in the non-monotonic effect of bisphenol-a on ca2+ entry in mouse pancreatic beta-cells. Sci. Rep. 2017, 7, 11770. [Google Scholar] [CrossRef]
- Soriano, S.; Alonso-Magdalena, P.; Garcia-Arevalo, M.; Novials, A.; Muhammed, S.J.; Salehi, A.; Gustafsson, J.A.; Quesada, I.; Nadal, A. Rapid insulinotropic action of low doses of bisphenol-A on mouse and human islets of Langerhans: Role of estrogen receptor beta. PLoS ONE 2012, 7, e31109. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Sun, X.; Qiu, L.; Wei, J.; Huang, Q.; Fang, C.; Ye, T.; Kang, M.; Shen, H.; Dong, S. Exposure to bisphenol A induces dysfunction of insulin secretion and apoptosis through the damage of mitochondria in rat insulinoma (INS-1) cells. Cell Death Dis. 2013, 4, e460. [Google Scholar] [CrossRef] [Green Version]
- Beydoun, H.A.; Khanal, S.; Zonderman, A.B.; Beydoun, M.A. Sex differences in the association of urinary bisphenol—A concentration with selected indices of glucose homeostasis among U.S. adults. Ann. Epidemiol. 2014, 24, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Chou, E.L.; Baecker, A.; You, N.C.; Song, Y.; Sun, Q.; Liu, S. Endocrine-disrupting chemicals, risk of type 2 diabetes, and diabetes-related metabolic traits: A systematic review and meta-analysis. J. Diabetes 2016, 8, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Martinez-Pinna, J.; Castellano-Munoz, M.; Dos Santos, R.S.; Medina-Gali, R.M.; Soriano, S.; Quesada, I.; Gustafsson, J.A.; Encinar, J.A.; Nadal, A. Bisphenol-S and Bisphenol-F alter mouse pancreatic beta-cell ion channel expression and activity and insulin release through an estrogen receptor ERbeta mediated pathway. Chemosphere 2021, 265, 129051. [Google Scholar] [CrossRef] [PubMed]
- Drzazga, A.; Kaminska, D.; Gliszczynska, A.; Gendaszewska-Darmach, E. Isoprenoid Derivatives of Lysophosphatidylcholines Enhance Insulin and GLP-1 Secretion through Lipid-Binding GPCRs. Int. J. Mol. Sci. 2021, 22, 5748. [Google Scholar] [CrossRef]
- Ranciere, F.; Botton, J.; Slama, R.; Lacroix, M.Z.; Debrauwer, L.; Charles, M.A.; Roussel, R.; Balkau, B.; Magliano, D.J.; D.E.S.I.R. Study Group. Exposure to Bisphenol A and Bisphenol S and Incident Type 2 Diabetes: A Case-Cohort Study in the French Cohort D.E.S.I.R. Environ. Health Perspect. 2019, 127, 107013. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Yao, Y.; Wang, B.; Han, L.; Wang, L.; Sun, H.; Chen, L. Association of urinary concentrations of bisphenols with type 2 diabetes mellitus: A case-control study. Environ. Pollut. 2018, 243, 1719–1726. [Google Scholar] [CrossRef]
- Bernardo, A.S.; Hay, C.W.; Docherty, K. Pancreatic transcription factors and their role in the birth, life and survival of the pancreatic beta cell. Mol. Cell. Endocrinol. 2008, 294, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Arevalo, M.; Alonso-Magdalena, P.; Servitja, J.M.; Boronat-Belda, T.; Merino, B.; Villar-Pazos, S.; Medina-Gomez, G.; Novials, A.; Quesada, I.; Nadal, A. Maternal Exposure to Bisphenol-A During Pregnancy Increases Pancreatic beta-Cell Growth During Early Life in Male Mice Offspring. Endocrinology 2016, 157, 4158–4171. [Google Scholar] [CrossRef]
- Wei, J.; Ding, D.; Wang, T.; Liu, Q.; Lin, Y. MiR-338 controls BPA-triggered pancreatic islet insulin secretory dysfunction from compensation to decompensation by targeting Pdx-1. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 5184–5195. [Google Scholar] [CrossRef] [Green Version]
- Rezg, R.; Abot, A.; Mornagui, B.; Knauf, C. Bisphenol S exposure affects gene expression related to intestinal glucose absorption and glucose metabolism in mice. Environ. Sci. Pollut. Res. Int. 2019, 26, 3636–3642. [Google Scholar] [CrossRef]
- Sun, X.; Lin, Y.; Huang, Q.; Shi, J.; Qiu, L.; Kang, M.; Chen, Y.; Fang, C.; Ye, T.; Dong, S. Di(2-ethylhexyl) phthalate-induced apoptosis in rat INS-1 cells is dependent on activation of endoplasmic reticulum stress and suppression of antioxidant protection. J. Cell. Mol. Med. 2015, 19, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, F.; Zhang, J.; Wang, K.; De, X.; Li, L.; Zhang, Y. Typical phthalic acid esters induce apoptosis by regulating the PI3K/Akt/Bcl-2 signaling pathway in rat insulinoma cells. Ecotoxicol. Environ. Saf. 2021, 208, 111461. [Google Scholar] [CrossRef]
- Ramachandran, V.; Arokia, V.A.M.; David, E.; Venkatachalam, K.; Vijayakumar, S.; Sankaran, V.; Balupillai, A.; Sangeetha, C.C.; Gothandam, K.M.; Kotakadi, V.S.; et al. Antidiabetic Activity of Gold Nanoparticles Synthesized Using Wedelolactone in RIN-5F Cell Line. Antioxidants 2019, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Wei, J.; Li, Y.; Chen, J.; Zhou, Z.; Song, L.; Wei, Z.; Lv, Z.; Chen, X.; Xia, W.; et al. Developmental exposure to di(2-ethylhexyl) phthalate impairs endocrine pancreas and leads to long-term adverse effects on glucose homeostasis in the rat. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E527–E538. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, P.; Balasubramanian, K. Gestational exposure to di(2-ethylhexyl) phthalate (DEHP) impairs pancreatic beta-cell function in F1 rat offspring. Toxicol. Lett. 2015, 232, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Venturelli, A.C.; Fischer, S.V.; de Morais, R.N.; Grassiolli, S.; Andrade, A.J.M. Effects of exposure to Di-(2-ethylhexyl) phthalate (DEHP) during lactation and puberty on sexual maturation and glycemic homeostasis in males rats. Clin. Nutr. ESPEN 2015, 10, e5–e12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shen, X.Y.; Zhang, W.W.; Chen, H.; Xu, W.P.; Wei, W. Di-(2-ethylhexyl) phthalate could disrupt the insulin signaling pathway in liver of SD rats and L02 cells via PPARgamma. Toxicol. Appl. Pharmacol. 2017, 316, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Zahid, K.R.; Wang, K.; Qian, Y.; Ma, P.; Ding, S.; Yang, X.; Wang, X. Adverse effect of DEHP exposure on the serum insulin level of Balb/c mice. Mol. Cell. Toxicol. 2016, 12, 83–91. [Google Scholar] [CrossRef]
- Committee, E.S.; More, S.; Benford, D.; Hougaard, B.S.; Bampidis, V.; Bragard, C.; Halldorsson, T.; Hernandez-Jerez, A.; Koutsoumanis, K.; Lambre, C.; et al. Opinion on the impact of non-monotonic dose responses on EFSA’s human health risk assessments. EFSA J. Eur. Food Saf. Auth. 2021, 19, e06877. [Google Scholar]
- Andrade, A.J.; Grande, S.W.; Talsness, C.E.; Grote, K.; Chahoud, I. A dose-response study following in utero and lactational exposure to di-(2-ethylhexyl)-phthalate (DEHP): Non-monotonic dose-response and low dose effects on rat brain aromatase activity. Toxicology 2006, 227, 185–192. [Google Scholar] [CrossRef]
- Do, R.P.; Stahlhut, R.W.; Ponzi, D.; Vom Saal, F.S.; Taylor, J.A. Non-monotonic dose effects of in utero exposure to di(2-ethylhexyl) phthalate (DEHP) on testicular and serum testosterone and anogenital distance in male mouse fetuses. Reprod. Toxicol. 2012, 34, 614–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maestri, L.; Negri, S.; Ferrari, M.; Ghittori, S.; Fabris, F.; Danesino, P.; Imbriani, M. Determination of perfluorooctanoic acid and perfluorooctanesulfonate in human tissues by liquid chromatography/single quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. RCM 2006, 20, 2728–2734. [Google Scholar] [CrossRef] [PubMed]
- Sant, K.E.; Jacobs, H.M.; Borofski, K.A.; Moss, J.B.; Timme-Laragy, A.R. Embryonic exposures to perfluorooctanesulfonic acid (PFOS) disrupt pancreatic organogenesis in the zebrafish, Danio rerio. Environ. Pollut. 2017, 220, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.P.; Cao, L.Y.; Li, C.H.; Guo, L.H.; Colbourne, J.; Ren, X.M. Perfluoroalkyl Substances Stimulate Insulin Secretion by Islet beta Cells via G Protein-Coupled Receptor 40. Environ. Sci. Technol. 2020, 54, 3428–3436. [Google Scholar] [CrossRef]
- Zhang, L.; Duan, X.; Sun, W.; Sun, H. Perfluorooctane sulfonate acute exposure stimulates insulin secretion via GPR40 pathway. Sci. Total Environ. 2020, 726, 138498. [Google Scholar] [CrossRef]
- Duan, X.; Sun, W.; Sun, H.; Zhang, L. Perfluorooctane sulfonate continual exposure impairs glucose-stimulated insulin secretion via SIRT1-induced upregulation of UCP2 expression. Environ. Pollut. 2021, 278, 116840. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.T.; Cheung, L.Y.; Chan, T.F.; Li, M.; Lai, K.P.; Wong, C.K.C. Characterization of PFOS toxicity on in-vivo and ex-vivo mouse pancreatic islets. Environ. Pollut. 2021, 289, 117857. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chen, P.C.; Lin, Y.C.; Lin, L.Y. Association among serum perfluoroalkyl chemicals, glucose homeostasis, and metabolic syndrome in adolescents and adults. Diabetes Care 2009, 32, 702–707. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.S.; Wen, L.L.; Chu, P.L.; Lin, C.Y. Association among total serum isomers of perfluorinated chemicals, glucose homeostasis, lipid profiles, serum protein and metabolic syndrome in adults: NHANES, 2013–2014. Environ. Pollut. 2018, 232, 73–79. [Google Scholar] [CrossRef]
- Cardenas, A.; Gold, D.R.; Hauser, R.; Kleinman, K.P.; Hivert, M.F.; Calafat, A.M.; Ye, X.; Webster, T.F.; Horton, E.S.; Oken, E. Plasma Concentrations of Per- and Polyfluoroalkyl Substances at Baseline and Associations with Glycemic Indicators and Diabetes Incidence among High-Risk Adults in the Diabetes Prevention Program Trial. Environ. Health Perspect. 2017, 125, 107001. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.C.; Glintborg, D.; Timmermann, C.A.G.; Nielsen, F.; Kyhl, H.B.; Andersen, H.R.; Grandjean, P.; Jensen, T.K.; Andersen, M. Perfluoroalkyl substances and glycemic status in pregnant Danish women: The Odense Child Cohort. Environ. Int. 2018, 116, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmermann, C.A.; Rossing, L.I.; Grontved, A.; Ried-Larsen, M.; Dalgard, C.; Andersen, L.B.; Grandjean, P.; Nielsen, F.; Svendsen, K.D.; Scheike, T.; et al. Adiposity and glycemic control in children exposed to perfluorinated compounds. J. Clin. Endocrinol. Metab. 2014, 99, E608–E614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, H.T.; Zhao, Y.G.; Leung, P.Y.; Wong, C.K. Perinatal exposure to perfluorooctane sulfonate affects glucose metabolism in adult offspring. PLoS ONE 2014, 9, e87137. [Google Scholar] [CrossRef] [PubMed]
- El Muayed, M.; Raja, M.R.; Zhang, X.; MacRenaris, K.W.; Bhatt, S.; Chen, X.; Urbanek, M.; O’Halloran, T.V.; Lowe, W.L., Jr. Accumulation of cadmium in insulin-producing beta cells. Islets 2012, 4, 405–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, G.A.; Chabosseau, P.; Bellomo, E.A.; Maret, W.; Mitchell, R.K.; Hodson, D.J.; Solomou, A.; Hu, M. Intracellular zinc in insulin secretion and action: A determinant of diabetes risk? Proc. Nutr. Soc. 2016, 75, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, H.; Xu, Y.; Xu, J.; Zhang, J.; Xi, Y.; Pi, H.; Yang, L.; Yu, Z.; Wu, Q.; Meng, Z.; et al. Cadmium exposure impairs pancreatic beta-cell function and exaggerates diabetes by disrupting lipid metabolism. Environ. Int. 2021, 149, 106406. [Google Scholar] [CrossRef]
- Chang, K.C.; Hsu, C.C.; Liu, S.H.; Su, C.C.; Yen, C.C.; Lee, M.J.; Chen, K.L.; Ho, T.J.; Hung, D.Z.; Wu, C.C.; et al. Cadmium induces apoptosis in pancreatic beta-cells through a mitochondria-dependent pathway: The role of oxidative stress-mediated c-Jun N-terminal kinase activation. PLoS ONE 2013, 8, e54374. [Google Scholar]
- Trevino, S.; Waalkes, M.P.; Hernandez, J.A.F.; Leon-Chavez, B.A.; Aguilar-Alonso, P.; Brambila, E. Chronic cadmium exposure in rats produces pancreatic impairment and insulin resistance in multiple peripheral tissues. Arch. Biochem. Biophys. 2015, 583, 27–35. [Google Scholar] [CrossRef]
- Sarmiento-Ortega, V.E.; Moroni-Gonzalez, D.; Diaz, A.; Eduardo, B.; Samuel, T. Oral Subacute Exposure to Cadmium LOAEL Dose Induces Insulin Resistance and Impairment of the Hormonal and Metabolic Liver-Adipose Axis in Wistar Rats. Biol. Trace Elem. Res. 2021. [Google Scholar] [CrossRef]
- Fitzgerald, R.; Olsen, A.; Nguyen, J.; Wong, W.; El Muayed, M.; Edwards, J. Pancreatic Islets Accumulate Cadmium in a Rodent Model of Cadmium-Induced Hyperglycemia. Int. J. Mol. Sci. 2020, 22, 360. [Google Scholar] [CrossRef]
- Li, X.; Li, M.; Xu, J.; Zhang, X.; Xiao, W.; Zhang, Z. Decreased Insulin Secretion but Unchanged Glucose Homeostasis in Cadmium-Exposed Male C57BL/6 Mice. J. Toxicol. 2019, 2019, 8121834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philibert, A.; Schwartz, H.; Mergler, D. An exploratory study of diabetes in a First Nation community with respect to serum concentrations of p,p′-DDE and PCBs and fish consumption. Int. J. Environ. Res. Public Health 2009, 6, 3179–3189. [Google Scholar] [CrossRef] [PubMed]
- Henriquez-Hernandez, L.A.; Luzardo, O.P.; Valeron, P.F.; Zumbado, M.; Serra-Majem, L.; Camacho, M.; Gonzalez-Antuna, A.; Boada, L.D. Persistent organic pollutants and risk of diabetes and obesity on healthy adults: Results from a cross-sectional study in Spain. Sci. Total Environ. 2017, 607–608, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Pavlikova, N.; Daniel, P.; Sramek, J.; Jelinek, M.; Sramkova, V.; Nemcova, V.; Balusikova, K.; Halada, P.; Kovar, J. Upregulation of vitamin D-binding protein is associated with changes in insulin production in pancreatic beta-cells exposed to p,p′-DDT and p,p′-DDE. Sci. Rep. 2019, 9, 18026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravassard, P.; Hazhouz, Y.; Pechberty, S.; Bricout-Neveu, E.; Armanet, M.; Czernichow, P.; Scharfmann, R. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J. Clin. Investig. 2011, 121, 3589–3597. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Abdulla, R.; Ferrero, H.; Soriano, S.; Boronat-Belda, T.; Alonso-Magdalena, P. Screening of Relevant Metabolism-Disrupting Chemicals on Pancreatic β-Cells: Evaluation of Murine and Human In Vitro Models. Int. J. Mol. Sci. 2022, 23, 4182. https://doi.org/10.3390/ijms23084182
Al-Abdulla R, Ferrero H, Soriano S, Boronat-Belda T, Alonso-Magdalena P. Screening of Relevant Metabolism-Disrupting Chemicals on Pancreatic β-Cells: Evaluation of Murine and Human In Vitro Models. International Journal of Molecular Sciences. 2022; 23(8):4182. https://doi.org/10.3390/ijms23084182
Chicago/Turabian StyleAl-Abdulla, Ruba, Hilda Ferrero, Sergi Soriano, Talía Boronat-Belda, and Paloma Alonso-Magdalena. 2022. "Screening of Relevant Metabolism-Disrupting Chemicals on Pancreatic β-Cells: Evaluation of Murine and Human In Vitro Models" International Journal of Molecular Sciences 23, no. 8: 4182. https://doi.org/10.3390/ijms23084182
APA StyleAl-Abdulla, R., Ferrero, H., Soriano, S., Boronat-Belda, T., & Alonso-Magdalena, P. (2022). Screening of Relevant Metabolism-Disrupting Chemicals on Pancreatic β-Cells: Evaluation of Murine and Human In Vitro Models. International Journal of Molecular Sciences, 23(8), 4182. https://doi.org/10.3390/ijms23084182