Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies

Abstract
:1. Introduction
Hepatocellular Carcinoma: Incidence, Risk Factors, Prognosis
2. Cancer Biology
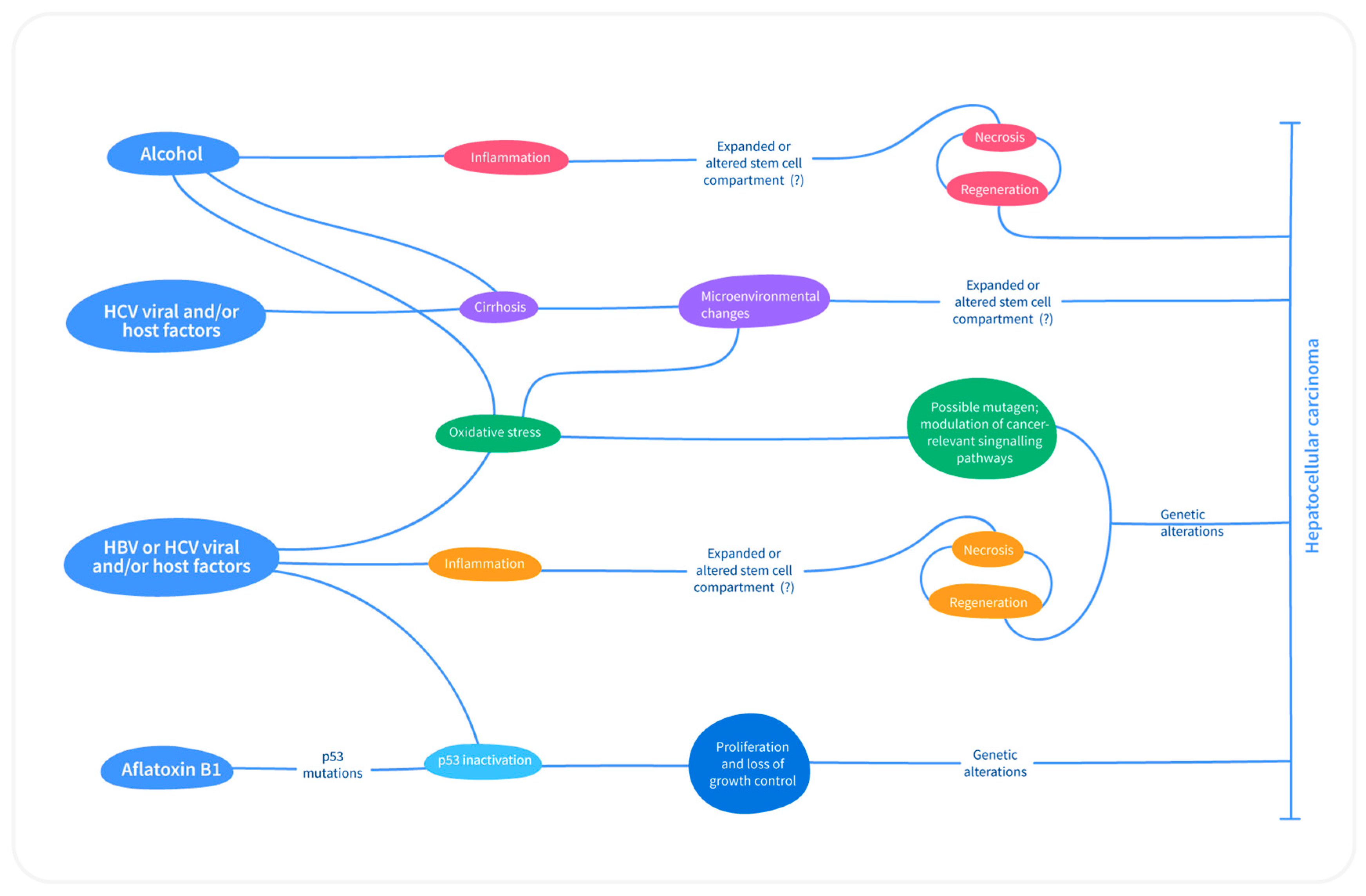
2.1. Fundamentals of Carcinogenesis
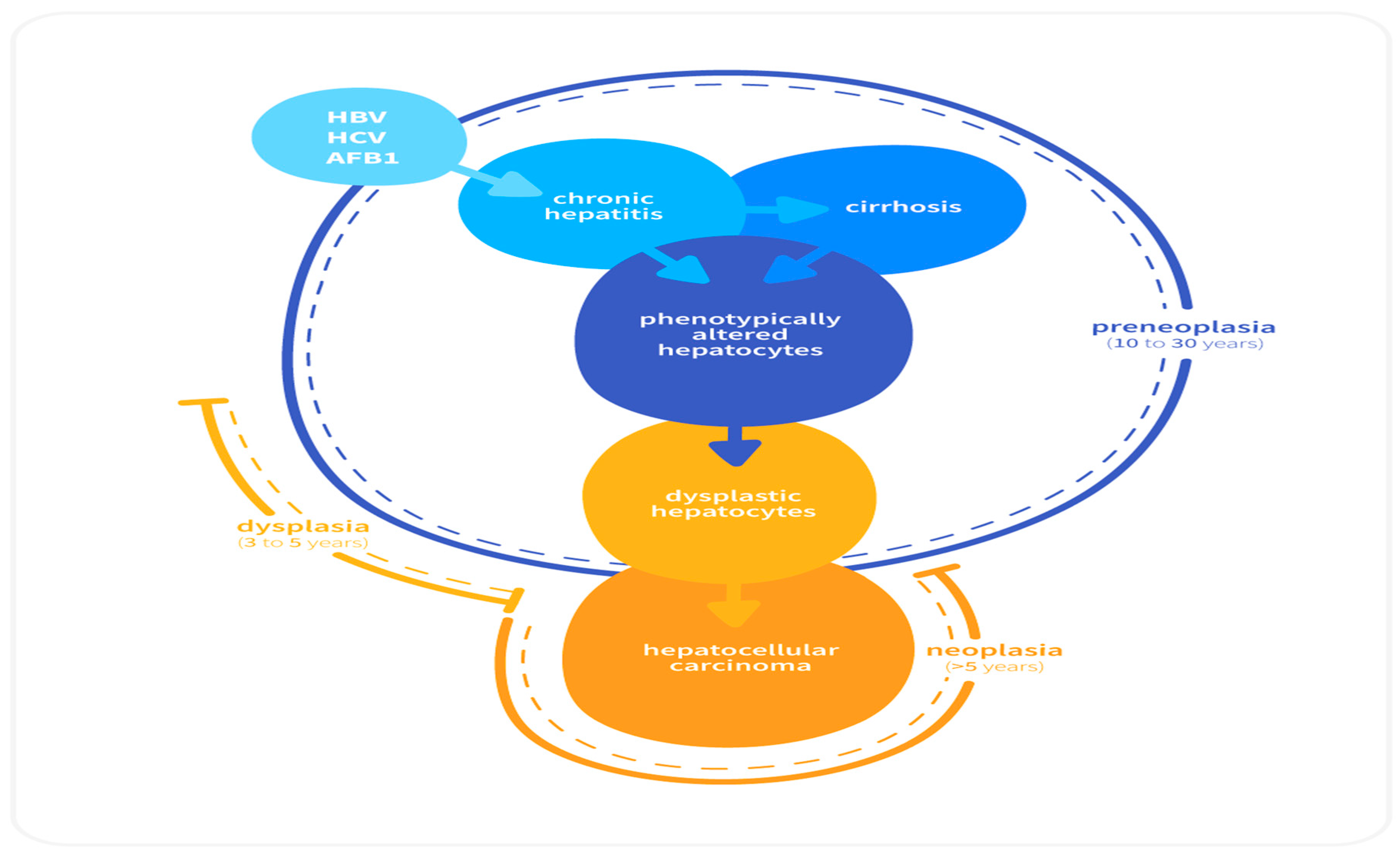
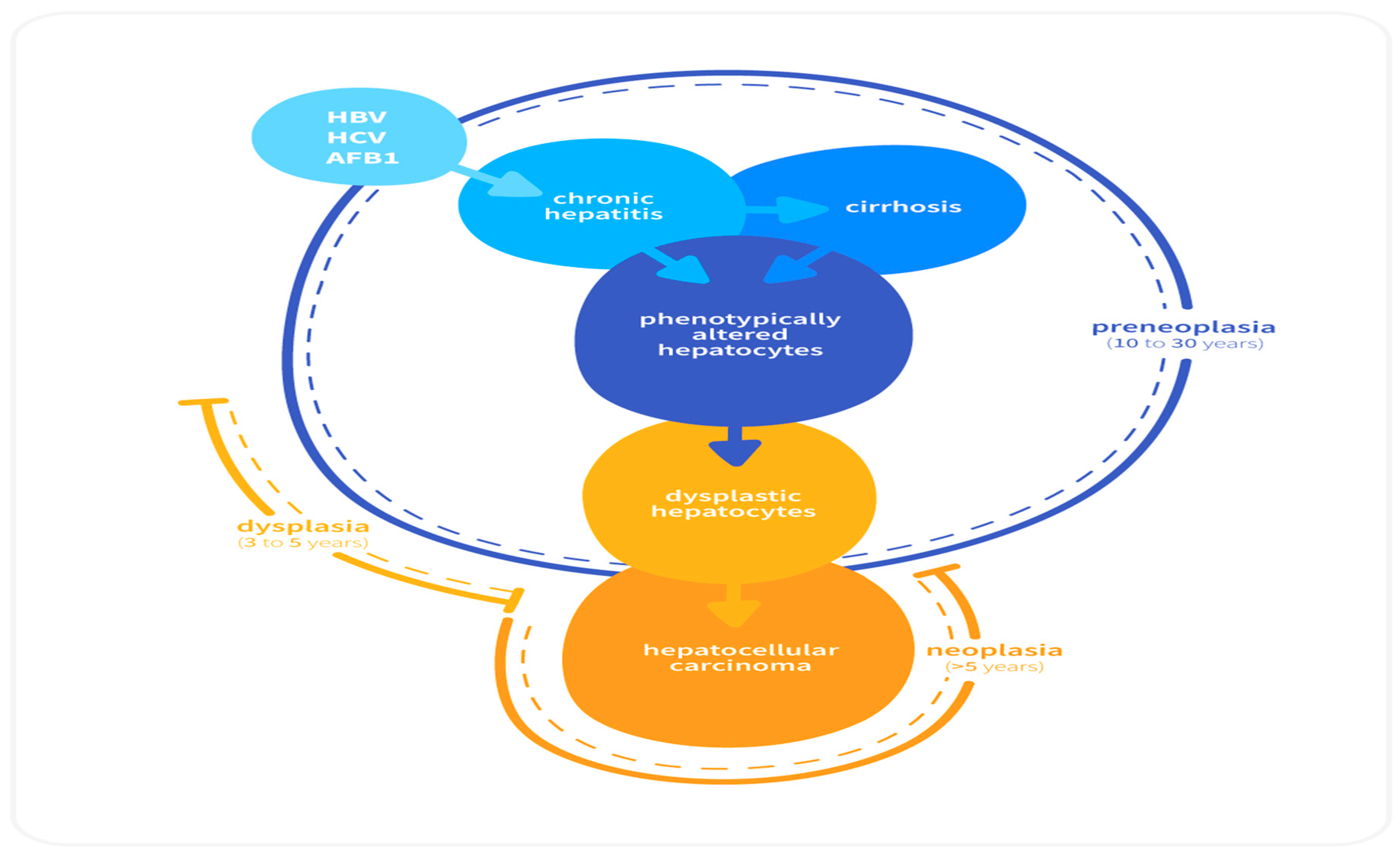
2.2. Sequential Development of HCC
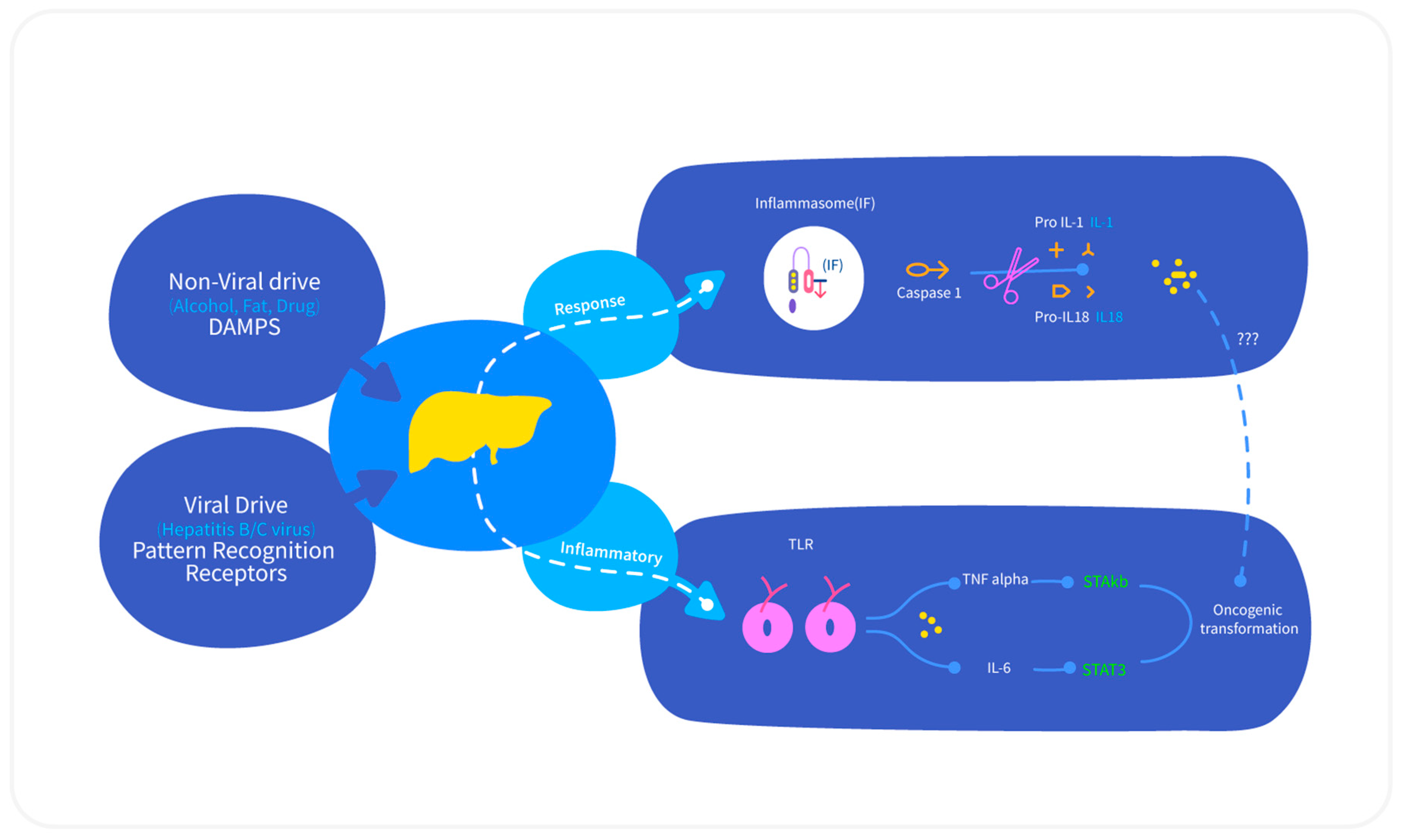
2.3. Role of Inflammation in HCC
2.3.1. IL-6 and TNF-α
2.3.2. Nuclear Factor-κβ (NF-κβ)
2.3.3. TGF-α
3. Molecular Events in HCC
- (i)
- Telomere shortening
- (ii)
- Copy number variants
- (iii)
- Single nucleotide variants and small deletions
- (iv)
- Epigenetic modifications
3.1. Telomere Shortening
3.2. Copy Number Variants
3.2.1. p53
3.2.2. pRb
3.2.3. Ras
3.2.4. c-myc
3.2.5. c-fos Activation
3.2.6. ErbB Receptor Family
3.2.7. Single Nucleotide Variants and Small Deletions
3.2.8. Epigenetic Alterations
3.3. Etiologic Factors and Associated Molecular Mechanisms in HCC
3.3.1. Viral Induced HCC
HBV Infection
HCV Infection
3.3.2. Nonalcoholic Fatty Liver Disease and HCC
3.3.3. Hemochromatosis and HCC
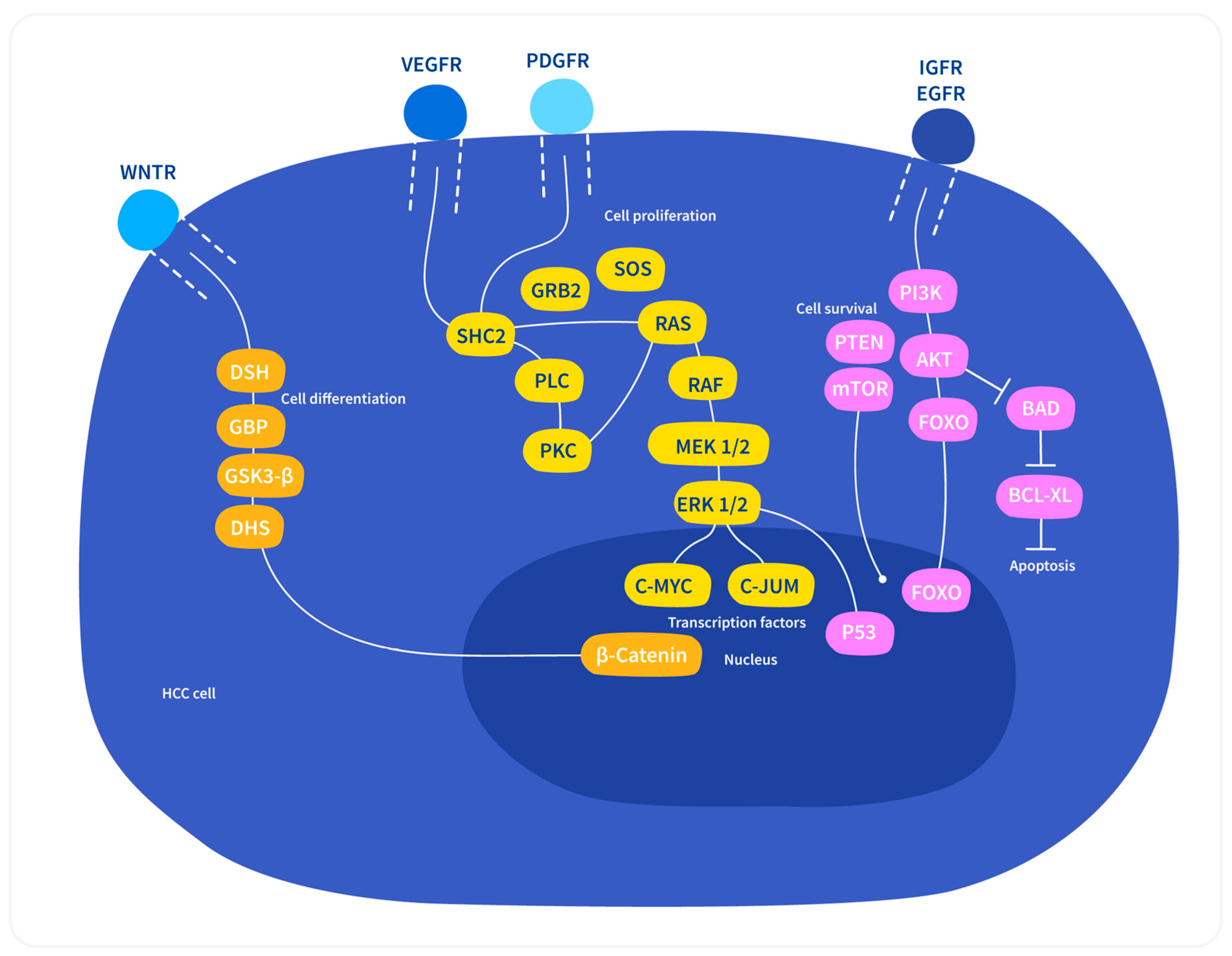
3.4. Different Cellular Signaling Pathways Linked to HCC
3.4.1. Wnt/β-Catenin Pathway
3.4.2. Ras/Raf/MAPK Pathway
3.4.3. PI3/AKT/mTOR Pathway
3.4.4. JAK/STAT Pathway
3.4.5. Ubiquitin-Proteasome (UP) Pathway
3.5. Angiogenesis and HCC
4. Molecular Targeted Therapies for HCC
4.1. Anti-Angiogenic Agents
4.1.1. Sorafenib
4.1.2. Lenvatinib
4.1.3. Regorafenib
4.1.4. Cabozantinib
4.2. EGFR Inhibitors
4.3. mTOR Inhibitors
4.4. c-MET Inhibitors
4.5. MEK Inhibitors
4.6. Other Molecular Targeted Agents
4.7. Immunotherapy for HCC Treatment
4.7.1. Pembrolizumab
4.7.2. Nivolumab
4.7.3. Bevacizumab
4.7.4. Ramucirumab
4.8. Immunotherapy in Adjuvant Setting
5. Challenges in Treatment of HCC Patients
6. Conclusions with Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Pan, H.; Fu, X.; Huang, W. Molecular mechanism of liver cancer. Anti-Cancer Agents Med. Chem. 2011, 11, 493–499. [Google Scholar] [CrossRef]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venook, A.P.; Papandreou, C.; Furuse, J.; De Guevara, L.L. The Incidence and Epidemiology of Hepatocellular Carcinoma: A Global and Regional Perspective. Oncologist 2010, 15 (Suppl. S4), 5–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Chen, K.F.; Chen, P.J. Treatment of liver cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a021535. [Google Scholar] [CrossRef] [PubMed]
- Golabi, P.; Fazel, S.; Otgonsuren, M.; Sayiner, M.; Locklear, C.T.; Younossi, Z.M. Mortality assessment of patients with hepatocellular carcinoma according to underlying disease and treatment modalities. Medicine 2017, 96, e5904. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tielent, J.; Jemal, A. Global cancer statistics. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina 2019, 55, 42. [Google Scholar] [CrossRef] [PubMed]
- Cha, C.; DeMatteo, R.P. Molecular mechanisms in hepatocellular carcinoma development. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Hamid, A.S.; Tesfamariam, I.G.; Zhang, Y.; Zhang, Z.G. Aflatoxin B1-induced hepatocellular carcinoma in developing countires: Geographical distribution, mechanism of action and prevention. Oncol. Lett. 2013, 5, 1087–1092. [Google Scholar] [CrossRef]
- Farazi, P.A.; Depinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R.N.; Steer, C.J.; Cressman, E.N.K. Molecular mechanisms of hepatocellular carcinoma. Hepatology 2008, 48, 2047–2063. [Google Scholar] [CrossRef] [PubMed]
- Raphael, S.W.; Yangde, Z.; Yuxiang, C. Hepatocellular Carcinoma: Focus on Different Aspects of Management. ISRN Oncol. 2012, 2012, 421673. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.R.; Gores, G.J. Hepatocellular Carcinoma: Molecular Pathways and New Therapeutic Targets. Semin. Liver Dis. 2005, 25, 212–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, L.; Rosenberg, D. Human Genes and Genomes, 1st ed.; Scientific Publishing Consultant: Lawrenceville, NJ, USA, 2012. [Google Scholar]
- Uma Devi, P. Basics of carcinogenesis. Health Adm. 1989, 17, 16–24. [Google Scholar]
- Chen, C.; Wang, G. Mechanisms of hepatocellular carcinoma and challenges and opportunities for molecular targeted therapy. World J. Hepatol. 2015, 7, 1964–1970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishna, G.; Rastogi, A.; Trehanpati, N.; Sen, B.; Khosla, R.; Sarin, S.K. From Cirrhosis to Hepatocellular Carcinoma: New Molecular Insights on Inflammation and Cellular Senescence. Liver Cancer 2013, 2, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A. The role of inflammation in liver cancer. In Book Inflammation and Cancer, Advances in Experimental Medicine and Biology; Aggarwal, B.B., Sung, B., Gupta, S.C., Eds.; Springer: Basel, Switzerland, 2014; pp. 401–435. [Google Scholar]
- Villanueva, A.; Luedde, T. The transition from inflammation to cancer in the liver. Clin. Liver Dis. 2016, 8, 89–93. [Google Scholar] [CrossRef]
- Yu, L.X.; Ling, Y.; Wang, H.Y. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precis. Oncol. 2018, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Sun, K.; Liu, W.; Sheng, D.; Zhao, S.; Gao, L.; Wei, L. Tumor necrosis factor-α promotes hepatocellular carcinogenesis through the activation of hepatic progenitor cells. Cancer Lett. 2018, 434, 22–32. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, W.L.; Li, Q.; Qiao, Q. Expression of transforming growth factor-α hepatitis and hepatitis B surface antigen in human hepatocellular carcinoma tissues and its significance. World J. Gastroenterol. 2004, 10, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Soukupova, J.; Malfettone, A.; Hyroššová, P.; Hernández-Alvarez, M.I.; Peñuelas-Haro, I.; Bertran, E.; Junza, A.; Capellades, J.; Giannelli, G.; Yanes, O.; et al. Role of the Transforming Growth Factor-β in regulating hepatocellular carcinoma oxidative metabolism. Sci. Rep. 2017, 7, 12486. [Google Scholar] [CrossRef] [PubMed]
- Bertino, G.; Demma, S.; Ardiri, A.; Proiti, M.; Malaguarnera, G.; Bertino, N.; Malaguarnera, M.; Malaguarnera, M. Hepatocellular Carcinoma: Novel Molecular Targets in Carcinogenesis for Future Therapies. BioMed Res. Int. 2014, 2014, 203693. [Google Scholar] [CrossRef] [PubMed]
- Merle, P.; Trepo, C. Molecular Mechanisms Underlying Hepatocellular Carcinoma. Viruses 2009, 1, 852–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M. Signaling Pathway and Molecular-Targeted Therapy for Hepatocellular Carcinoma. Dig. Dis. 2011, 29, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Güller, M.; Toualbi-Abed, K.; Legrand, A.; Michel, L.; Mauviel, A.; Bernuau, D.; Daniel, F. c-Fos overexpression increases the proliferation of human hepatocytes by stabilizing nuclear Cyclin D1. World J. Gastroenterol. 2008, 14, 6339–6346. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Yoon, S.K.; Lencioni, R. The Etiology of Hepatocellular Carcinoma and Consequences for Treatment. Oncologist 2010, 15 (Suppl. S4), 14–22. [Google Scholar] [CrossRef] [Green Version]
- Siu, L.S.; Foont, J.F.; Wands, J.R. Hepatitis C virus and alcohol. Semin. Liver Dis. 2009, 29, 188–199. [Google Scholar] [CrossRef]
- Pianko, S.; Patella, S.; Sievert, W. Alcohol consumption induces hepatocyte apoptosis in patients with chronic hepatitis C infection. J. Gastroenterol. Hepatol. 2000, 15, 798–805. [Google Scholar] [CrossRef]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis-(NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Kaseb, A.O.; Tsimberidou, A.M.; Wolff, R.A.; Kurzrock, R. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget 2014, 5, 3012–3022. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004, 127, S79–S86. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.P. Hepatocellular carcinoma and the ubiquitin–proteasome system. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Stotz, M.; Gerger, A.; Haybaeck, J.; Kiesslich, T.; Bullock, M.D.; Pichler, M. Molecular Targeted Therapies in Hepatocellular Carcinoma: Past, Present and Future. Anticancer Res. 2015, 35, 5737–5744. [Google Scholar]
- Tanaka, S.; Arii, S. Molecular Targeted Therapies in Hepatocellular Carcinoma. Semin. Oncol. 2012, 39, 486–492. [Google Scholar] [CrossRef]
- Shen, Y.C.; Lin, Z.Z.; Hsu, C.H.; Hsu, C.; Shao, Y.Y.; Cheng, A.L. Clinical Trials in Hepatocellular Carcinoma: An Update. Liver Cancer 2013, 2, 345–364. [Google Scholar] [CrossRef]
- Kudo, M. Lenvatinib May Drastically Change the Treatment Landscape of Hepatocellular Carcinoma. Liver Cancer 2018, 7, 1–19. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.M.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III renadomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Johnson, P.J.; Qin, S.; Park, J.W.; Poon, R.T.; Raoul, J.L.; Philip, P.A.; Hsu, C.H.; Hu, T.H.; Heo, J.; Xu, J.; et al. Brivanib Versus Sorafenib As First-Line Therapy in Patients With Unresectable, Advanced Hepatocellular Carcinoma: Results From the Randomized Phase III BRISK-FL Study. J. Clin. Oncol. 2013, 31, 3517–3524. [Google Scholar] [CrossRef] [PubMed]
- Cainip, C.; Qin, S.; Huang, W.T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.K.; Chen, P.J.; Toh, H.C.; et al. Linifanib versus Sorafenib in patients with advanced hepatocellular carcinoma: Results of randomized phase III trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- FDA Approves Lenvatinib for Unresectable Hepatocellular Carcinoma. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm617185.htm (accessed on 12 April 2019).
- Cerrito, L.; Ponziani, F.R.; Garcovich, M.; Tortora, A.; Annicchiarico, B.E.; Pompili, M.; Siciliano, M.; Gasbarrini, A. Regorafenib: A promising treatment for hepatocellular carcinoma. Expert Opin. Pharmacother. 2018, 19, 1941–1948. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- FDA Approves Cabozanitab for Hepatocellular Carcinoma. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm629512.htm (accessed on 17 April 2019).
- Chua, C.W.L.; Choo, S.P. Targeted Therapy in Hepatocellular Carcinoma. Int. J. Hepatol. 2011, 2011, 348297. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kudo, M.; Assenat, E.; Cattan, S.; Kang, Y.K.; Lim, H.Y.; Poon, R.T.; Blanc, J.F.; Vogel, A.; Chen, C.L.; et al. Effect of Everolimus on survival in advanced hepatocellular carcinoma after failure of Sorafenib The EVOLVE-1 Randomized Clinical Trial. JAMA 2014, 312, 57–67. [Google Scholar] [CrossRef]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Caremoli, E.R.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- Tai, W.M.; Yong, W.P.; Lim, C.; Low, L.S.; Tham, C.K.; Koh, T.S.; Ng, Q.S.; Wang, W.W.; Wang, L.Z.; Hartano, S.; et al. A phase Ib study of selumetinib (AZD6244, ARRY-142886) in combination with sorafenib in advanced hepatocellular carcinoma (HCC). Ann. Oncol. 2016, 27, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Okusaka, T.; Ueno, H.; Ikeda, M.; Takezako, Y.; Morizane, C. Phase I study of TAC-101, an oral synthetic retinoid, in Japanese patients with advanced hepatocellular carcinoma. Cancer Sci. 2012, 103, 1524–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phase 2 Study of TAC-101 Combined with Transcatheter Arterial Chemoembolization (TACE) Versus TACE alone in Japanese Patients with Advanced Hepatocellular Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00667628 (accessed on 18 April 2019).
- Tovoli, F.; Negrini, G.; Benevento, F.; Faggiano, C.; Goio, E.; Granito, A. Systemic treatments for hepatocellular carcinoma: Challenges and future perspectives. Hepatic Oncol. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Finn, R.S.; Cattan, S.; Edeline, J.; Ogasawara, S.; Palmer, D.H.; Verslype, C.; Zagonel, V.; Rosmorduc, O.; Vogel, A.; et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J. Clin. Oncol. 2018, 36, 209. [Google Scholar] [CrossRef]
- Finn, R.S.; Chan, S.L.; Zhu, A.X.; Knox, J.J.; Cheng, A.L.; Siegel, A.B.; Bautista, O.; Watson, P.; Kudo, M. KEYNOTE-240: Randomized phase III study of pembrolizumab versus best supportive care for second-line advanced hepatocellular carcinoma. J. Clin. Oncol. 2017, 35 (Suppl. S4). [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Sorafenib as First-Line Treatment in Patients with Advanced Hepatocellular Carcinoma (CheckMate 459: Checkpoint Pathway and Nivolumab Clinical Trial Evaluation 459 an Investigational Immuno-Therapy Study of Nivolumab Compared to Sorafenib as a First Treatment in Patients with Advanced Hepatocellular Carcinoma. Available online: https://ichgcp.net/clinical-trials-registry/NCT02576509 (accessed on 5 June 2019).
- Zhu, A.X.; Kang, Y.K.; Yen, C.J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomized, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- FDA Approves Ramucirumab for Hepatocellular Carcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ramucirumab-hepatocellular-carcinoma (accessed on 5 June 2019).
- Brown, Z.J.; Greten, T.F.; Heinrich, B. Adjuvant Treatment of Hepatocellular Carcinoma: Prospect of Immunotherapy. Hepatology 2019. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.H.; Lim, Y.S.; Yeon, J.E.; Song, T.J.; Yu, S.J. Sustained efficacy of adjuvant immunotherapy with cytokine-induced killer cells for hepatocellular carcinoma: An extended 5-year follow-up. Cancer Immunol. Immunother. 2019, 68, 23–32. [Google Scholar] [CrossRef]
- Okusaka, T.; Ikeda, M. Immunotherapy for hepatocellular carcinoma: Current status and future perspectives. ESMO Open 2018, 3, e000455. [Google Scholar] [CrossRef] [PubMed]
- Colagrande, S.; Inghilesi, A.L.; Aburas, S.; Taliani, G.G.; Nardi, C.; Marra, F. Challenges of advanced hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 7645–7659. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, M.; Daniele, B.; Pignata, S.; Gallo, C.; De Maio, E.; Morabito, A.; Piccirillo, M.C.; Perrone, F. Is human hepatocellular carcinoma a hormone-responsive tumor? World J. Gastroenterol. 2008, 14, 1682–1689. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Target | Trial Phase, Name, clinicaltrial.gov Number | OS | TTP | Results | Ref. |
|---|---|---|---|---|---|---|
| Sorafenib | Multi-kinase inhibitor | Phase III vs. placebo (SHARP; NCT00105443) | 10.7 months vs. 7.9 months (p < 0.001) | 4.1 months vs. 4.9 months (p = 0.77) | Positive | [43,44] |
| Phase III vs. placebo (Asia-Pacific; NCT00492752) | 6.5 months vs. 4.2 months (p = 0.014) | 2.8 months vs. 4.9 months (p = 0.0005) | ||||
| Sunitinib | VEGFR, PDGFR inhibitor | Phase III vs. sorafenib (SUN 1170; NCT00699374) | 8.1 months vs. 10.2 months (two-sided p = 0.019) | 4.1 months vs. 3.8 months (two-sided p = 0.3082) | Negative | [41,42] |
| Linifanib | VEGFR, PDGFR inhibitor | Phase III vs. sorafenib (LIGHT; NCT01009593) | 9.1 months vs. 9.8 months (p = NS) | 5.4 months vs. 4.0 months (p = 0.001) | Negative | [41,42,46] |
| Brivanib | VEGFR, PDGFR, FGFR inhibitor | Phase III vs. sorafenib (BRISK-FL; NCT00858871) | 9.5 months vs. 9.9 months (p = 0.3116) | 4.2 months vs. 4.1 months (p = 0.853) | Negative | [41,42] |
| Lenvatinib | Multi-kinase inhibitor | Phase III vs. sorafenib (REFLECT; NCT01761266) | 13.6 months vs. 12.3 months | 8.9 months vs. 3.7 months (p < 0.0001) | Positive | [47] |
| Regorafenib | Multi-kinase inhibitor | Phase III vs. placebo (RESORCE; NCT01774344) | 10.6 months vs. 7.8 months (p < 0.0001) | - | Positive | [50] |
| Cabozantinib | Multi-kinase inhibitor | Phase III vs. placebo (CELESTIAL; NCT01908426) | 10.2 months vs. 8.0 months (p = 0.005) | - | Positive | [51] |
| Erlotinib | EGFR inhibitor | Phase III vs. sorafenib (SEARCH; NCT00901901 | 9.5 months vs. 8.5 months (p > 0.05) | 3.2 months vs. 4.0 months (p > 0.05) | Negative | [41] |
| Everolimus | mTOR inhibitor | Phase III vs. placebo (EVOLVE-1; NCT01035229) | 7.6 months vs. 7.3 months | 3.0 months vs. 2.6 months | Negative | [54] |
| Trivantinib | c-Met inihibitor | Phase III vs. placebo (NCT01755767) | 8.4 months vs. 9.1 months (p = 0.81) | - | Negative | [55] |
| Selumetinib | MEK inhibitor | Phase II vs. placebo (NCT00604721) | - | - | - | [41] |
| Phase Ib vs. sorafenib | 14.4 months with selumetinib | - | ||||
| Pembrolizumab | Anti-PD-1 monoclonal antibody | Phase II (KEYNOTE-224; NCT02702414) | 12.9 months | 4.9 months | Negative | [61] |
| Phase III vs. placebo (KEYNOTE-240; NCT02702401) | - | - | ||||
| Bevacizumab | Anti-VEGF antibody | Phase II vs. placebo | 53% (1 year) 28% (2 years) 23% (3 years) | - | Negative | [53] |
| Bevacizumab + Gemcitabine + Oxaliplatin | Phase II | 9.6 months | - | Negative | [53] | |
| Nivolumab | Anti-PD-1 monoclonal antibody | Phase I/II (CheckMate-040; NCT01658878) | 15.0 months | 3.4 months | Positive | [62] |
| Ramucirumab | Human IgG1 monoclonal antibody | Phase III vs. placebo (REACH-2; NCT02435433) | 8.5 months vs. 7.3 months (p = 0.0199) | Median time to radiologic progression 3.0 months vs. 1.6 months (p < 0.0001) | Positive | [64] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqahtani, A.; Khan, Z.; Alloghbi, A.; S. Said Ahmed, T.; Ashraf, M.; M. Hammouda, D. Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies. Medicina 2019, 55, 526. https://doi.org/10.3390/medicina55090526
Alqahtani A, Khan Z, Alloghbi A, S. Said Ahmed T, Ashraf M, M. Hammouda D. Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies. Medicina. 2019; 55(9):526. https://doi.org/10.3390/medicina55090526
Chicago/Turabian StyleAlqahtani, Ali, Zubair Khan, Abdurahman Alloghbi, Tamer S. Said Ahmed, Mushtaq Ashraf, and Danae M. Hammouda. 2019. "Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies" Medicina 55, no. 9: 526. https://doi.org/10.3390/medicina55090526
APA StyleAlqahtani, A., Khan, Z., Alloghbi, A., S. Said Ahmed, T., Ashraf, M., & M. Hammouda, D. (2019). Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies. Medicina, 55(9), 526. https://doi.org/10.3390/medicina55090526