

Combined aCGH and Exome Sequencing Analysis Improves Autism Spectrum Disorders Diagnosis: A Case Report
 , and
, and
Abstract
:1. Introduction
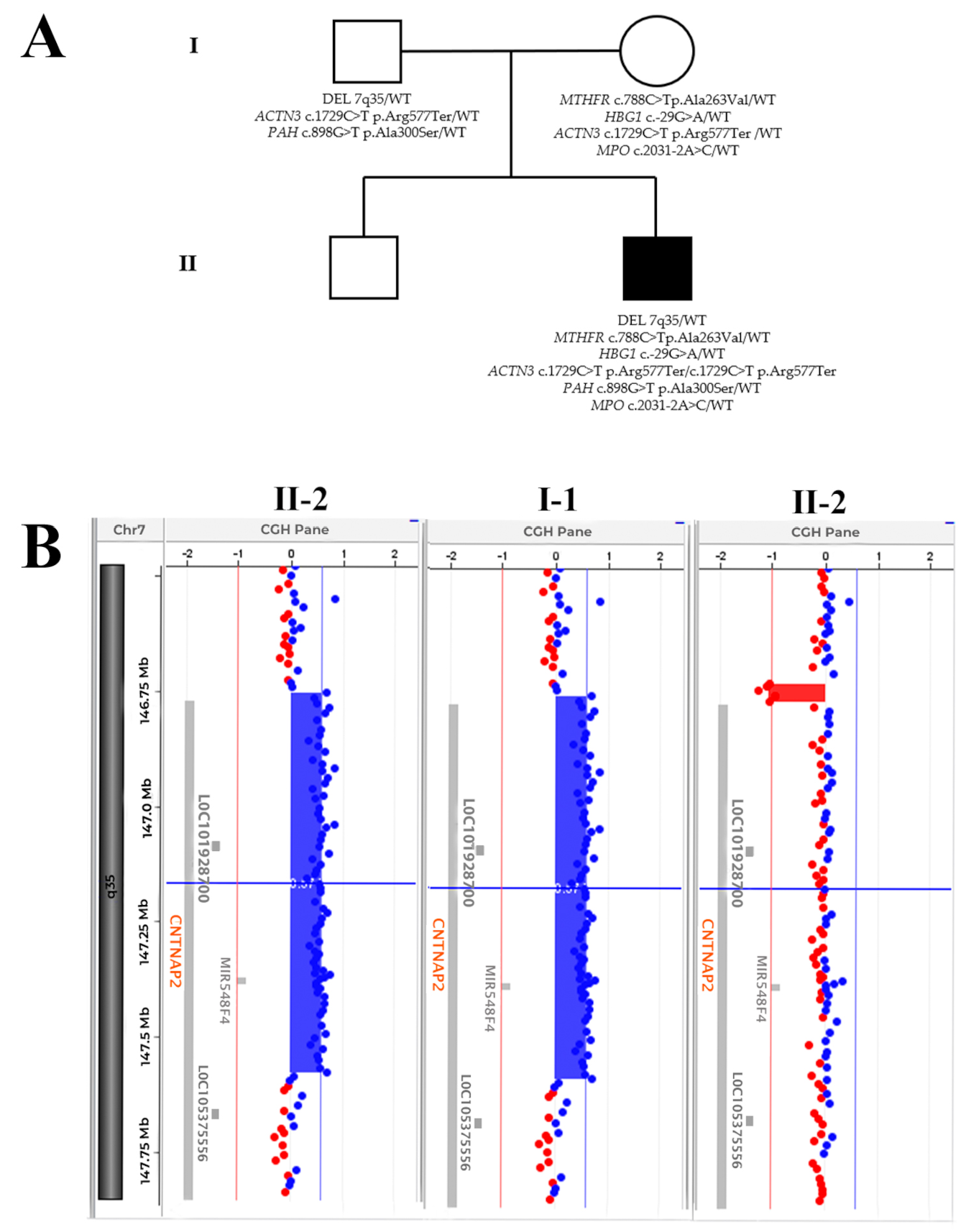
2. Case Report
2.1. Case Presentation
2.2. Molecular Analyses
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al-Dewik, N.; Al-Jurf, R.; Styles, M.; Tahtamouni, S.; Alsharshani, D.; Alsharshani, M.; Ahmad, A.I.; Khattab, A.; Al Rifai, H.; Walid Qoronfleh, M. Overview and Introduction to Autism Spectrum Disorder (ASD). Adv. Neurobiol. 2020, 24, 3–42. [Google Scholar]
- Lovrečić, L.; Rajar, P.; Volk, M.; Bertok, S.; Gnidovec Stražišar, B.; Osredkar, D.; Jekovec Vrhovšek, M.; Peterlin, B. Diagnostic Efficacy and New Variants in Isolated and Complex Autism Spectrum Disorder Using Molecular Karyotyping. J. Appl. Genet. 2018, 59, 179–185. [Google Scholar]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and Epigenetics of Autism Spectrum Disorder—Current Evidence in the Field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar]
- Lord, C.; Brugha, T.S.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.J.H.; Jones, R.M.; Pickles, A.; State, M.W.; et al. Autism Spectrum Disorder. Nat. Rev. Dis. Prim. 2020, 6, 5. [Google Scholar]
- Egger, G.; Roetzer, K.M.; Noor, A.; Lionel, A.C.; Mahmood, H.; Schwarzbraun, T.; Boright, O.; Mikhailov, A.; Marshall, C.R.; Windpassinger, C.; et al. Identification of Risk Genes for Autism Spectrum Disorder through Copy Number Variation Analysis in Austrian Families. Neurogenetics 2014, 15, 117–127. [Google Scholar]
- Rosti, R.O.; Sadek, A.A.; Vaux, K.K.; Gleeson, J.G. The Genetic Landscape of Autism Spectrum Disorders. Dev. Med. Child Neurol. 2014, 56, 12–18. [Google Scholar]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The Familial Risk of Autism. JAMA 2014, 311, 1770–1777. [Google Scholar]
- Brancaccio, M.; Mennitti, C.; Cesaro, A.; Monda, E.; D’argenio, V.; Casaburi, G.; Mazzaccara, C.; Ranieri, A.; Fimiani, F.; Barretta, F.; et al. Multidisciplinary In-Depth Investigation in a Young Athlete Suffering from Syncope Caused by Myocardial Bridge. Diagnostics 2021, 11, 2144. [Google Scholar]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic Analysis Resolves Differential Diagnosis of a Familial Syndromic Dilated Cardiomyopathy: A New Case of Alström Syndrome. Mol. Genet. Genom. Med. 2020, 8, e1260. [Google Scholar]
- Nunziato, M.; Starnone, F.; Lombardo, B.; Pensabene, M.; Condello, C.; Verdesca, F.; Carlomagno, C.; De Placido, S.; Pastore, L.; Salvatore, F.; et al. Fast Detection of a BRCA2 Large Genomic Duplication by next Generation Sequencing as a Single Procedure: A Case Report. Int. J. Mol. Sci. 2017, 18, 2487. [Google Scholar]
- Iossa, S.; Costa, V.; Corvino, V.; Auletta, G.; Barruffo, L.; Cappellani, S.; Ceglia, C.; Cennamo, G.; D’Adamo, A.P.; D’Amico, A.; et al. Phenotypic and Genetic Characterization of a Family Carrying Two Xq21.1-21.3 Interstitial Deletions Associated with Syndromic Hearing Loss. Mol. Cytogenet. 2015, 8, 18. [Google Scholar]
- Sanna, V.; Ceglia, C.; Tarsitano, M.; Lombardo, B.; Coppola, A.; Zarrilli, F.; Castaldo, G.; Di Minno, G. Aberrant F8 Gene Intron 1 Inversion with Concomitant Duplication and Deletion in a Severe Hemophilia A Patient from Southern Italy. J. Thromb. Haemost. 2013, 11, 195–197. [Google Scholar]
- Zebisch, A.; Schulz, E.; Grosso, M.; Lombardo, B.; Acierno, G.; Sill, H.; Iolascon, A. Identification of a Novel Variant of Epsilon-Gamma-Delta-Beta Thalassemia Highlights Limitations of next Generation Sequencing. Am. J. Hematol. 2015, 90, E52–E54. [Google Scholar]
- Mozzillo, E.; Cozzolino, C.; Genesio, R.; Melis, D.; Frisso, G.; Orrico, A.; Lombardo, B.; Fattorusso, V.; Discepolo, V.; Della Casa, R.; et al. Mulibrey Nanism: Two Novel Mutations in a Child Identified by Array CGH and DNA Sequencing. Am. J. Med. Genet. Part A 2016, 170, 2196–2199. [Google Scholar]
- De Angelis, C.; Nardelli, C.; Concolino, P.; Pagliuca, M.; Setaro, M.; De Paolis, E.; De Placido, P.; Forestieri, V.; Scaglione, G.L.; Ranieri, A.; et al. Case Report: Detection of a Novel Germline PALB2 Deletion in a Young Woman with Hereditary Breast Cancer: When the Patient’s Phenotype History Doesn’t Lie. Front. Oncol. 2021, 11, 602523. [Google Scholar]
- Iafusco, F.; De Sanctis, P.; Pirozzi, D.; Capone, S.; Lombardo, B.; Gambale, A.; Confetto, S.; Zanfardino, A.; Iolascon, A.; Pastore, L.; et al. Molecular Diagnosis of MODY3 Permitted to Reveal a de Novo 12q24.31 Deletion and to Explain a Complex Phenotype in a Young Diabetic Patient. Clin. Chem. Lab. Med. 2019, 57, e306–e310. [Google Scholar]
- Di Stefano, C.; Lombardo, B.; Fabbricatore, C.; Munno, C.; Caliendo, I.; Gallo, F.; Pastore, L. Oculo-Facio-Cardio-Dental (OFCD) Syndrome: The First Italian Case of BCOR and Co-Occurring OTC Gene Deletion. Gene 2015, 559, 203–206. [Google Scholar]
- Lombardo, B.; Zarrilli, F.; Ceglia, C.; Vitale, A.; Keller, S.; Sarchiapone, M.; Carli, V.; Stuppia, L.; Chiariotti, L.; Castaldo, G.; et al. Two Novel Genomic Rearrangements Identified in Suicide Subjects Using A-CGH Array. Clin. Chem. Lab. Med. 2015, 53, e245–e248. [Google Scholar]
- Lombardo, B.; Ceglia, C.; Verdesca, F.; Vitale, A.; Perrotta, C.; Leggiero, E.; Pastore, L. CGH Array for the Identification of a Compound Heterozygous Mutation in the CYP1B1 Gene in a Patient with Bilateral Anterior Segment Dysgenesis. Clin. Chem. Lab. Med. 2019, 57, e63–e66. [Google Scholar]
- Vitale, A.; Labruna, G.; Mancini, A.; Alfieri, A.; Iaffaldano, L.; Nardelli, C.; Pasanisi, F.; Pastore, L.; Buono, P.; Lombardo, B. 3q29 Microduplication in a Small Family with Complex Metabolic Phenotype from Southern Italy. Clin. Chem. Lab. Med. 2018, 56, e167–e170. [Google Scholar]
- Lombardo, B.; Ceglia, C.; Tarsitano, M.; Pierucci, I.; Salvatore, F.; Pastore, L. Identification of a Deletion in the NDUFS4 Gene Using Array-Comparative Genomic Hybridization in a Patient with Suspected Mitochondrial Respiratory Disease. Gene 2014, 535, 376–379. [Google Scholar]
- Tarsitano, M.; Ceglia, C.; Novelli, A.; Capalbo, A.; Lombardo, B.; Pastore, L.; Fioretti, G.; Vicari, L.; Pisanti, M.A.; Friso, P.; et al. Microduplications in 22q11.2 and 8q22.1 Associated with Mild Mental Retardation and Generalized Overgrowth. Gene 2014, 536, 213–216. [Google Scholar]
- Rodenas-Cuadrado, P.; Ho, J.; Vernes, S.C. Shining a Light on CNTNAP2: Complex Functions to Complex Disorders. Eur. J. Hum. Genet. 2014, 22, 171–178. [Google Scholar]
- Belloso, J.M.; Bache, I.; Guitart, M.; Caballin, M.R.; Halgren, C.; Kirchhoff, M.; Ropers, H.H.; Tommerup, N.; Tümer, Z. Disruption of the CNTNAP2 Gene in a t(7;15) Translocation Family without Symptoms of Gilles de La Tourette Syndrome. Eur. J. Hum. Genet. 2007, 15, 711–713. [Google Scholar]
- Toma, C.; Pierce, K.D.; Shaw, A.D.; Heath, A.; Mitchell, P.B.; Schofield, P.R.; Fullerton, J.M. Comprehensive Cross-Disorder Analyses of CNTNAP2 Suggest It Is Unlikely to Be a Primary Risk Gene for Psychiatric Disorders. PLoS Genet. 2018, 14, e1007535. [Google Scholar]
- Xiaoping, L.; Zhengmao, H.; Yiqun, H.; Zhimin, X.; Zhigao, L.; Yu, P.; Fengxiao, B.; Jie, L.; Guanglei, X.; Xiaoyun, M.; et al. Association Analysis of CNTNAP2 Polymorphisms with Autism in the Chinese Han Population. Psychiatr. Genet. 2010, 20, 113–117. [Google Scholar]
- Peñagarikano, O.; Abrahams, B.S.; Herman, E.I.; Winden, K.D.; Gdalyahu, A.; Dong, H.; Sonnenblick, L.I.; Gruver, R.; Almajano, J.; Bragin, A.; et al. Absence of CNTNAP2 Leads to Epilepsy, Neuronal Migration Abnormalities, and Core Autism-Related Deficits. Cell 2011, 147, 235–246. [Google Scholar]
- Nascimento, P.P.; Bossolani-Martins, A.L.; Rosan, D.B.A.; Mattos, L.C.; Brandão-Mattos, C.; Fett-Conte, A.C. Single Nucleotide Polymorphisms in the CNTNAP2 Gene in Brazilian Patients with Autistic Spectrum Disorder. Genet. Mol. Res. 2016, 15, 1–8. [Google Scholar]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism Spectrum Disorder: Neuropathology and Animal Models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar]
- Scala, M.; Anijs, M.; Battini, R.; Madia, F.; Capra, V.; Scudieri, P.; Verrotti, A.; Zara, F.; Minetti, C.; Vernes, S.C.; et al. Hyperkinetic Stereotyped Movements in a Boy with Biallelic CNTNAP2 Variants. Ital. J. Pediatr. 2021, 47, 1–5. [Google Scholar]
- Zhou, W.Z.; Zhang, J.; Li, Z.; Lin, X.; Li, J.; Wang, S.; Yang, C.; Wu, Q.; Ye, A.Y.; Wang, M.; et al. Targeted Resequencing of 358 Candidate Genes for Autism Spectrum Disorder in a Chinese Cohort Reveals Diagnostic Potential and Genotype–Phenotype Correlations. Hum. Mutat. 2019, 40, 801–815. [Google Scholar]
- Friedman, J.I.; Vrijenhoek, T.; Markx, S.; Janssen, I.M.; Van Der Vliet, W.A.; Faas, B.H.W.; Knoers, N.V.; Cahn, W.; Kahn, R.S.; Edelmann, L.; et al. CNTNAP2 Gene Dosage Variation Is Associated with Schizophrenia and Epilepsy. Mol. Psychiatry 2008, 13, 261–266. [Google Scholar]
- Pickering, C.; Kiely, J. ACTN3: More than Just a Gene for Speed. Front. Physiol. 2017, 8, 1080. [Google Scholar]
- North, K.N.; Yang, N.; Wattanasirichaigoon, D.; Mills, M.; Easteal, S.; Beggs, A.H. A Common Nonsense Mutation Results in α-Actinin-3 Deficiency in the General Population. Nat. Genet. 1999, 21, 353–354. [Google Scholar]
- Houweling, P.J.; Papadimitriou, I.D.; Seto, J.T.; Pérez, L.M.; Del Coso, J.; North, K.N.; Lucia, A.; Eynon, N. Is Evolutionary Loss Our Gain? The Role of ACTN3 p.Arg577Ter (R577X) Genotype in Athletic Performance, Ageing, and Disease. Hum. Mutat. 2018, 39, 1774–1787. [Google Scholar]
- Liew, S.C.; Gupta, E. Das Methylenetetrahydrofolate Reductase (MTHFR) C677T Polymorphism: Epidemiology, Metabolism and the Associated Diseases. Eur. J. Med. Genet. 2015, 58, 1–10. [Google Scholar]
- Sibarov, D.A.; Boikov, S.I.; Karelina, T.V.; Antonov, S.M. GluN2 Subunit-Dependent Redox Modulation of NMDA Receptor Activation by Homocysteine. Biomolecules 2020, 10, 1441. [Google Scholar]
- Rajagopal, S.; Fitzgerald, A.A.; Deep, S.N.; Paul, S.; Poddar, R. Role of GluN2A NMDA Receptor in Homocysteine-Induced Prostaglandin E2 Release from Neurons. J. Neurochem. 2019, 150, 44–55. [Google Scholar]
- Deep, S.N.; Mitra, S.; Rajagopal, S.; Paul, S.; Poddar, R. GluN2A-NMDA Receptor-Mediated Sustained Ca2+ Influx Leads to Homocysteine-Induced Neuronal Cell Death. J. Biol. Chem. 2019, 294, 11154–11165. [Google Scholar]
- Poddar, R.; Chen, A.; Winter, L.; Rajagopal, S.; Paul, S. Role of AMPA Receptors in Homocysteine-NMDA Receptor-Induced Crosstalk between ERK and P38 MAPK. J. Neurochem. 2017, 142, 560–573. [Google Scholar]
- Chisholm, K.; Lin, A.; Abu-Akel, A.; Wood, S.J. The Association between Autism and Schizophrenia Spectrum Disorders: A Review of Eight Alternate Models of Co-Occurrence. Neurosci. Biobehav. Rev. 2015, 55, 173–183. [Google Scholar]

{kind=link}
| Chr | Gene | cDNA * | Protein * | Reference SNP ID | Status | Inheritance | Associated Phenotype † | Clinvar Classification | ACMG/AMP § Classification |
|---|---|---|---|---|---|---|---|---|---|
| 1 | MTHFR | c.788C>T | p.Ala263Val | rs1801133 | Het | F | Homocystinuria due to MTHFR deficiency—AR Neural tube defects, susceptibility to—AR Schizophrenia, susceptibility to—AD Thromboembolism, susceptibility to—AD Vascular disease, susceptibility to—AD | Drug response | VUS |
| 11 | HBG1 | c.-29G>A | rs368698783 | Het | F | Fetal hemoglobin quantitative trait locus 1—AD | Pathogenetic | Benign | |
| 11 | ACTN3 | c.1729C>T | p.Arg577Ter | rs1815739 | Hom | F + M | Alpha-actinin-3 deficiency—AR Sprinting performance—AR | VUS | VUS |
| 12 | PAH | c.898G>T | p.Ala300Ser | rs5030853 | Het | M | Phenylketonuria AR | Pathogenetic | Pathogenetic |
| 17 | MPO | c.2031-2A>C | rs35897051 | Het | F | Myeloperoxidase deficiency—AR Alzheimer’s disease, susceptibility to—AD | Pathogenetic | Pathogenetic |
| cDNA * | Protein * | Reference SNP ID | Status | Inheritance | Clinvar Classification | ACMG/AMP § Classification |
|---|---|---|---|---|---|---|
| c.1897+25A>G | rs2074715 | Het | M | Benign | Benign | |
| c.2099-53C>A | rs2538962 | Het | F | na | Benign | |
| c.3382-34G>A | rs3801976 | Hom | F + M | na | Benign | |
| c.3716-12_3716-9dupCTTT | rs142426153 | Hom | F | VUS | VUS | |
| c.3716-6C>G | rs77025884 | Het | F | Benign | Benign | |
| c.3723G>A | p.Ala1241= | rs9648691 | Het | F | Benign | Benign |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranieri, A.; Veneruso, I.; La Monica, I.; Pascale, M.G.; Pastore, L.; D’Argenio, V.; Lombardo, B. Combined aCGH and Exome Sequencing Analysis Improves Autism Spectrum Disorders Diagnosis: A Case Report. Medicina 2022, 58, 522. https://doi.org/10.3390/medicina58040522
Ranieri A, Veneruso I, La Monica I, Pascale MG, Pastore L, D’Argenio V, Lombardo B. Combined aCGH and Exome Sequencing Analysis Improves Autism Spectrum Disorders Diagnosis: A Case Report. Medicina. 2022; 58(4):522. https://doi.org/10.3390/medicina58040522
Chicago/Turabian StyleRanieri, Annaluisa, Iolanda Veneruso, Ilaria La Monica, Maria Grazia Pascale, Lucio Pastore, Valeria D’Argenio, and Barbara Lombardo. 2022. "Combined aCGH and Exome Sequencing Analysis Improves Autism Spectrum Disorders Diagnosis: A Case Report" Medicina 58, no. 4: 522. https://doi.org/10.3390/medicina58040522