
Coffin-Lowry Syndrome Induced by RPS6KA3 Gene Variation in China: A Case Report in Twins

Abstract
:1. Introduction
2. Case Report
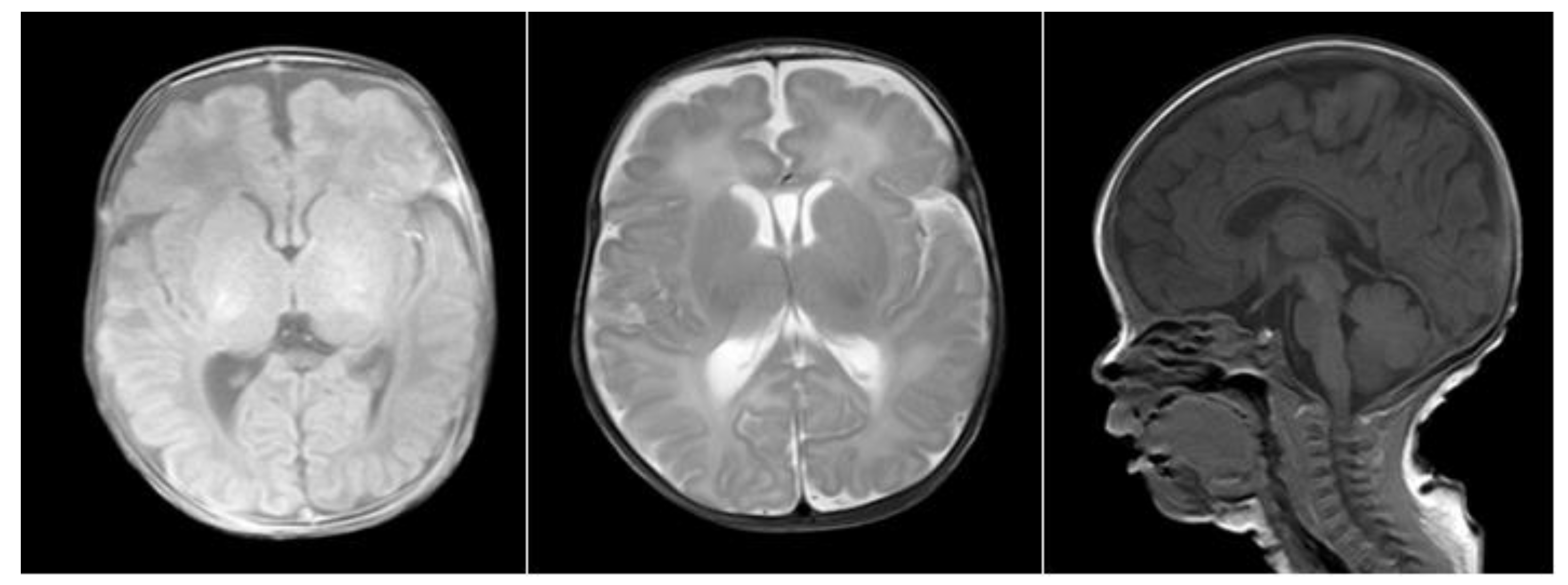
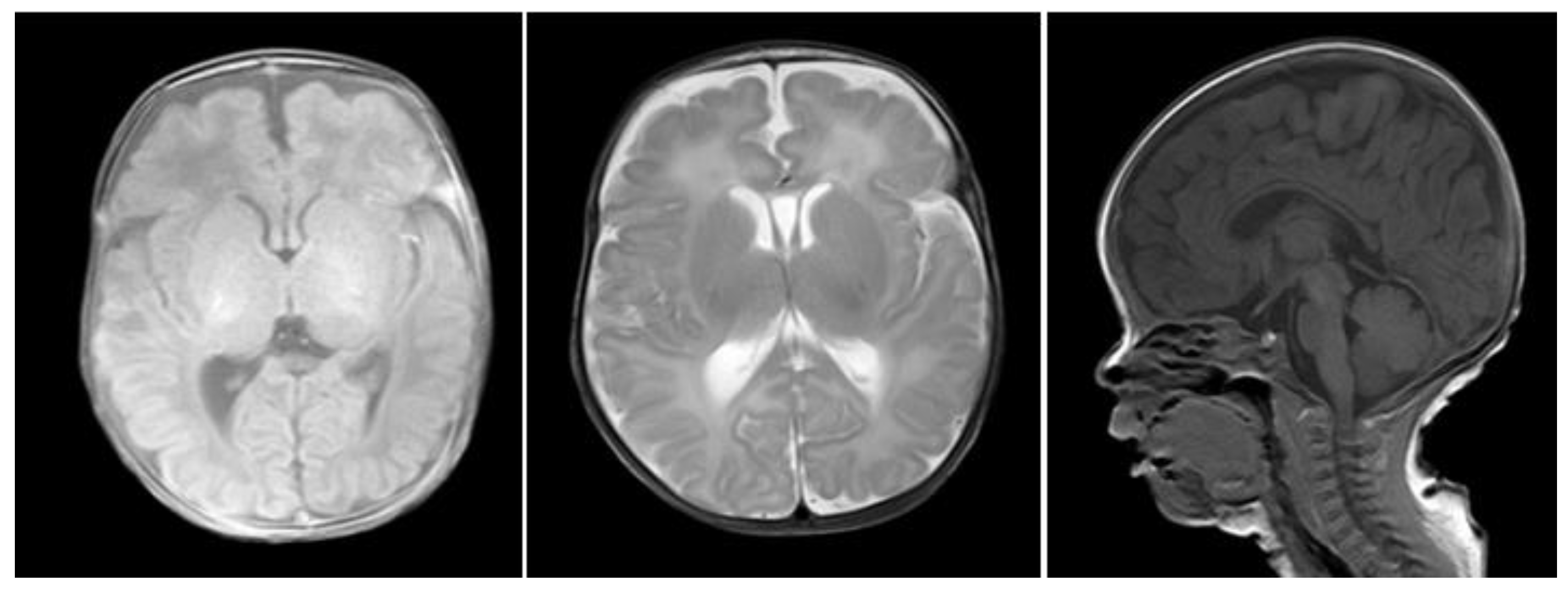
2.1. Clinical Data
2.2. Experimental Methods
2.2.1. Test Sample Collection
2.2.2. Experimental Process
2.3. Sequencing Results
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rogers, R.C.; Abidi, F.E. Coffin-Lowry Syndrome; GeneReviews: Seattle, WA, USA, 2018. [Google Scholar]
- Hanauer, A.; Young, I.D. Coffin-Lowry syndrome: Clinical and molecular features. J. Med. Genet. 2002, 39, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, Y.; Wang, Z. Coffin-Lowry syndrome: A case study. Chin. J. Pediatr. 2006, 44, 1. [Google Scholar]
- Wang, Y.; Meng, Y.; Liang, S.; Shi, H.; Zha, S. Genetic diagnosis of Coffin-Lowry syndrome. In Proceedings of the 8th National Conference on Medical Genetics, Heilongjiang, China, 1 July 2009. [Google Scholar]
- Zhang, L.; Cao, Y.; Zhang, G.; Zhang, L. Coffin-Lowry syndrome: A case report and literature review. Clin. Pediatr. 2018, 36, 265–267. [Google Scholar]
- Shen, N.; Liu, Y.; Zhang, K.; Lv, Y.; Gao, M.; Ma, J.; Xu, L.; Gai, Z. Analysis of genetic variation in a Coffin-Lowry syndrome family. Chin. J. Med. Genet. 2019, 8, 798–800. [Google Scholar]
- Liu, Y.; Zeng, Y.; Liu, L.; Huang, Y.; Yu, L.; Ding, H. Molecular diagnosis and family genetic analysis of Coffin-Lowry syndrome induced by RPS6KA3 gene variation. Chin. J. Prenat. Diagn. 2021, 13, 34–39. [Google Scholar]
- Li, Q.; Su, W.; Liu, X.; Zhang, G. Coffin-Lowry syndrome with hypopigmentation spots: A case study. Chin. J. Dermatol. 2015, 4, 79–80. [Google Scholar]
- Fung, J.L.; Rethanavelu, K.; Luk, H.M.; Ho, M.S.; Lo, I.F.; Chung, B.H. Coffin-Lowry syndrome in Chinese. Am. J. Med. Genet. Part A 2019, 179, 2043–2048. [Google Scholar] [CrossRef]
- Delaunoy, J.P.; Dubos, A.; Marques Pereira, P.; Hanauer, A. Identification of novel mutations in the RSK2 gene (RPS6KA3) in patients with Coffin-Lowry syndrome. Clin. Genet. 2006, 70, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Tos, T.; Alp, M.Y.; Aksoy, A.; Ceylaner, S.; Hanauer, A. A familial case of Coffin-Lowry syndrome caused by RPS6KA3 C.898C>T mutation associated with multiple abnormal brain imaging findings. Genet. Couns 2015, 26, 47–52. [Google Scholar] [PubMed]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Zhu, L.; Zheng, J.; Wu, D.; Shao, J. Growth concerns in Coffin-Lowry syndrome: A case report and literature review. Front. Pediatr. 2019, 6, 430. [Google Scholar] [CrossRef] [PubMed]
- Yamoto, K.; Saitsu, H.; Fujisawa, Y.; Kato, F.; Matsubara, K.; Fukami, M.; Kagami, M.; Ogata, T. Coffin Lowry syndrome in a girl with 46,XX,t(X;11)(p22;p15)dn: Identification of RPS6KA3 disruption by whole genome sequencing. Case Rep. 2020, 8, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Touma Boulos, M.; Moukarzel, A.; Yammine, T.; Salem, N.; Souaid, M.; Farra, C. Novel missense mutation c.1784A>G, p.Tyr595Cys in RPS6KA3 gene responsible for Coffin–Lowry syndrome in a family with variable features and diabetes 2. Clin. Dysmorphol. 2021, 30, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Rojnueangnit, K.; Jones, J.R.; Basehore, M.J.; Robin, N.H. Classic phenotype of Coffin-lowry syndrome in a female with stimulus-induced drop episodes and a genotype with preserved N-terminal kinase domain. Am. J. Med. Genet. Part A 2014, 164, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Martinez, H.R.; Niu, M.C.; Sutton, V.R.; Pignatelli, R.; Vatta, M.; Jefferies, J.L. Coffin-Lowry syndrome and left ventricular noncompaction cardiomyopathy with a restrictive. Am. J. Med. Genet. Part A 2011, 155, 3030–3034. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, M.; Foletti, G.; Baumer, A.; Bottani, A.; Novy, J. Recurrent Nonconvulsive Status Epilepticus in a Patient with Coffin-Lowry Syndrome. Mol. Syndromol. 2015, 6, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Chromosome Position | Transcript Number | Exon/Intron | Nucleotide Change | Amino Acid Change | Heterozygous/ Homozygous | Related Diseases | Inheritance Mode | Variation Classification | Source of Variation |
|---|---|---|---|---|---|---|---|---|---|---|
| RPS6KA3 | chrX: 20195150 | NM_004586.3 | Exon 11 | c.898C>T | p.R300* | Hemizygous | Coffin-Lowry syndrome | XLD and XLD | Pathogenic | De novo mutation |
| The Sample Name | Sample Number | Test Data Volume (bp) | Mean Sequencing Depth | Target Area Coverage | Proportion of Coverage Area over 10× | Proportion of Coverage Area over 20× |
|---|---|---|---|---|---|---|
| Proband | WES22030470 | 17376448800 | 220.48 | 99.97% | 99.90% | 99.77% |
| Father | WES22030471 | 19205799600 | 244.88 | 99.96% | 99.90% | 99.78% |
| Mother | WES22030472 | 14236229100 | 181.27 | 99.82% | 99.69% | 99.48% |
| Gene | Chromosome Position | Transcript Number | Exon/Intron | Nucleotide Change | Amino Acid Change | Heterozygous/ Homozygous | Variation Classification | Source of Variation |
|---|---|---|---|---|---|---|---|---|
| RPS6KA3 | chrX: 20195150 | NM_004586.3 | Exon 11 | c.898C>T | p.R300* | Hemizygous | Pathogenic | De novo mutation |
| Case | Age of Diagnosis | Gender | Special Facial Features | Tapered Fingers | Developmental Delay | Hypotonia | Abnormal Hearing | Cranial MRI Abnormalities | RPS6KA3 Mutation | |
|---|---|---|---|---|---|---|---|---|---|---|
| This study | 1 | 5M | M | + | - | + | + | + | + | Exon 11:c.898C>T (p.R300*) |
| 2 | 5M | M | + | - | + | + | + | + | Exon 11:c.898C>T (p.R300*) | |
| Li, Y [3] | 3 | 2Y6M | M | + | + | + | + | N/A | + | N/A |
| Wang, Y [4] | 4 | 13Y | M | + | + | + | + | N/A | N/A | c.r889_890delAG |
| Zhang, L [5] | 5 | 4Y3M | M | + | + | + | + | - | - | Exon 5:c.340C>T |
| Shen, N [6] | 6 | 2Y | M | + | + | + | + | + | - | Exon12:c.966_967delAA(p.Arg323Thr fs*11) |
| Liu, Y [7] | 7 | 1Y | M | + | - | + | + | + | N/A | C.1672cC> T (p.R558*) |
| 8 | 4Y | M | + | - | + | + | + | + | Hemizygote variation(c.325+2_325+3insT) | |
| Li, Q [8] | 9 | 3Y4M | M | + | + | + | + | - | - | Exon 19:c.1841+1G>A |
| Fung, J.L [9] | 10 | 10Y | M | + | + | + | + | - | N/A | Deletion of exon 9 and 10 |
| 11 | 36Y | F | + | + | N/A | N/A | - | N/A | Deletion of exon 9 and 10 | |
| 12 | 3Y | M | + | + | + | + | + | N/A | Exon17: c.1449T>A, p.(Tyr483Ter) | |
| 13 | 19Y | F | + | + | + | + | - | N/A | Exon 17: c.1449T>A, p.(Tyr483Ter | |
| 14 | 42Y | F | + | + | + | N/A | - | N/A | Exon 17: c.1449T>A, p.(Tyr483Ter) | |
| 15 | 2Y | M | + | + | + | + | - | N/A | c.1842-1G>T | |
| 16 | 25Y | M | + | + | + | + | - | N/A | Exon 16: c.1428_ 1430delTAT | |
| 17 | 18Y | M | + | + | + | + | - | N/A | Exon 9: c.638G>A, p.(Gly213Asp) | |
| 18 | 16Y | F | + | + | + | - | - | N/A | Exon 7: c.501delA, p.(Glu168fsTer14) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Li, H.; Qiang, S. Coffin-Lowry Syndrome Induced by RPS6KA3 Gene Variation in China: A Case Report in Twins. Medicina 2022, 58, 958. https://doi.org/10.3390/medicina58070958
Jin H, Li H, Qiang S. Coffin-Lowry Syndrome Induced by RPS6KA3 Gene Variation in China: A Case Report in Twins. Medicina. 2022; 58(7):958. https://doi.org/10.3390/medicina58070958
Chicago/Turabian StyleJin, Huiying, Haifeng Li, and Shu Qiang. 2022. "Coffin-Lowry Syndrome Induced by RPS6KA3 Gene Variation in China: A Case Report in Twins" Medicina 58, no. 7: 958. https://doi.org/10.3390/medicina58070958
APA StyleJin, H., Li, H., & Qiang, S. (2022). Coffin-Lowry Syndrome Induced by RPS6KA3 Gene Variation in China: A Case Report in Twins. Medicina, 58(7), 958. https://doi.org/10.3390/medicina58070958