
Mass Spectrometry-Based Proteomics: Analyses Related to Drug-Resistance and Disease Biomarkers

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Discussion
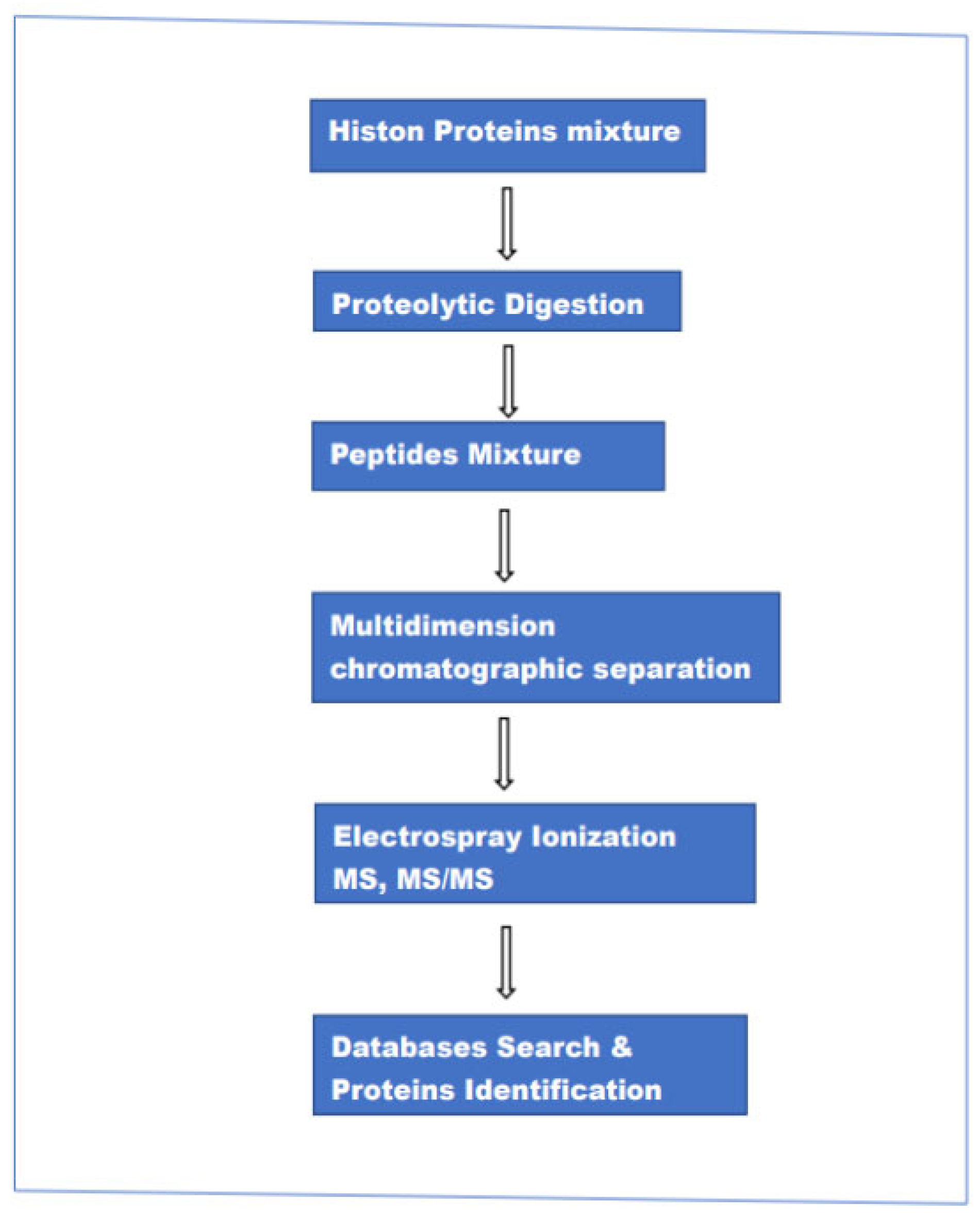
2.1. MS-Based Methods for the Analyses of Histone Proteins
2.2. Shotgun Analyses
Enhancement of MS/MS in Top/Middle-Down Methods
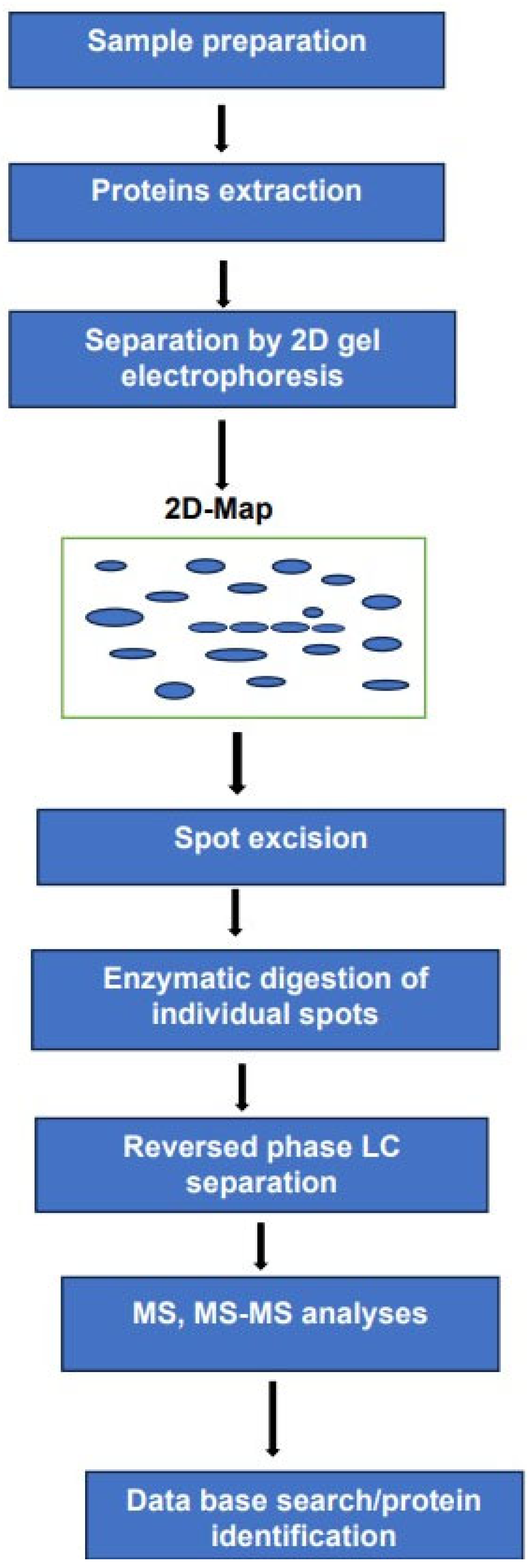
2.3. Two-Dimensional Gel Electrophoresis 2DE
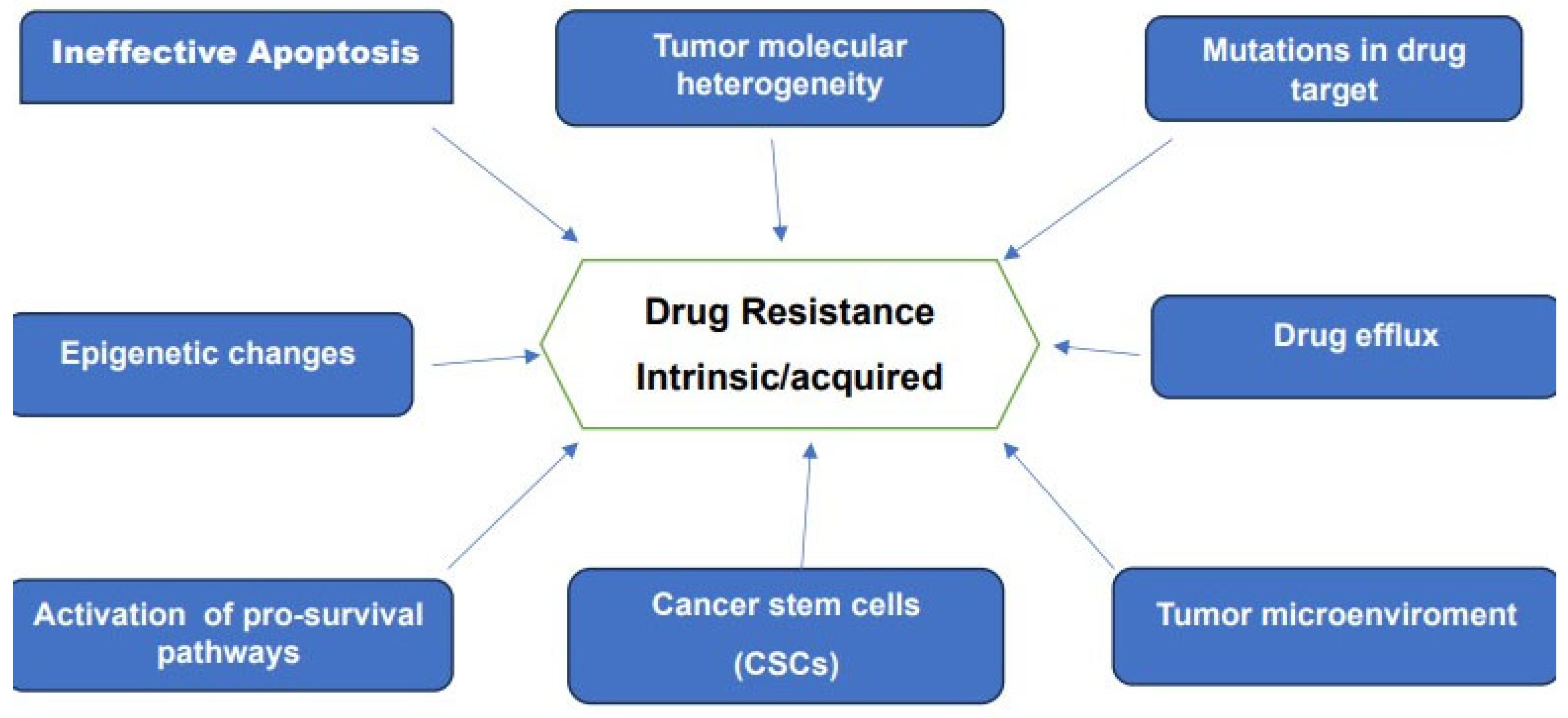
2.4. Drug Resistance in Cancer
2.5. MS Investigation of Proteins Implicated in Drugs Efflux
2.6. Single-Cell MS to Investigate Acquired Drug Resistance
2.7. Drug Resistance in Childhood Cancers
3. Potential Epigenetic Biomarkers Based on MS Analyses
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| UVPD | Ultraviolet photo dissociation |
| IM-MS | Ion mobility mass spectrometry |
| LIT | Lineariontrap |
| ICR | Ion cyclotron resonance |
| TOF | Time offlight |
| PTMs | Post translational modifications |
| LC/MS-MS | Liquid chromatography/tandem mass spectrometry |
| SILAC | amino acids in cell culture |
| MDR | Multidrug resistance |
| MB | Medulloblastoma |
| CSCs | Cancer stem cells |
| MSI | Mass spectrometry imaging |
| MALDI | Matrix assisted laser desorption ionization |
| ALL | Acute lymphoblastic leukemia |
| CRC | Colorectal cancer |
| ELISA | Enzyme-linked immunosorbent assay |
| 2DE | Two-Dimensional Gel Electrophoresis |
| ECD | Electron capture dissociation |
| ETD | electron transfer dissociation |
| CID | collision-induced dissociation |
| AA | Amino acids |
| PI | Isoelectric point |
| Mr | Relative molecular weight |
| TMT | Tandem mass tag |
| SCX | Strong cation exchange chromatography |
| WCX | Weak cation exchange |
| MRM | Multiple reaction monitoring |
References
- Lai, W.K.M.; Pugh, B.F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol. 2017, 18, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012, 13, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Ramesh, V.; Locasale, J.W. Metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 2020, 21, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; Bannister, A.J. Two genomes, one cell: Mitochondrial–nuclear coordination via epigenetic pathways. Mol. Metab. 2020, 38, 100942. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef]
- Cosgrove, M.S.; Boeke, J.D.; Wolberger, C. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 2004, 11, 1037–1043. [Google Scholar] [CrossRef]
- Neumann, H.; Hancock, S.M.; Buning, R.; Routh, A.; Chapman, L.; Somers, J.; Owen-Hughes, T.; van Noort, J.; Rhodes, D.; Chin, J.W. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol. Cell 2009, 36, 153–163. [Google Scholar] [CrossRef]
- Ramazi, S.; Allahverdi, A.; Zahiri, J. Evaluation of post-translational modifications in histone proteins: A review on histone modification defects in developmental and neurological disorders. J. Biosci. 2020, 45, 135. [Google Scholar] [CrossRef]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ’oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Wang, N.; Ma, T.; Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancer. Signal Transduct. Target. Ther. 2023, 8, 69. [Google Scholar] [CrossRef]
- Hayashi, T.; Konishi, I. Correlation of anti-tumour drug resistance with epigenetic regulation. Br. J. Cancer 2021, 124, 681–682. [Google Scholar] [CrossRef]
- Soler-Botija, C.; Gálvez-Montón, C.; Bayés-Genís, A. Epigenetic Biomarkers in Cardiovascular Diseases. Front Genet. 2019, 10, 950. [Google Scholar] [CrossRef]
- Kamińska, K.; Nalejska, E.; Kubiak, M.; Wojtysiak, J.; Żołna, L.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Epigenetic Biomarkers in Oncology. Mol. Diagn. Ther. 2019, 23, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, A.T. The Promise and the Problems of Epigenetics Biomarkers in Cancer. Expert Opin. Med. Diagn. 2011, 5, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Egelhofer, T.A.; Minoda, A.; Klugman, S.; Lee, K.; Kolasinska-Zwierz, P.; Alekseyenko, A.A.; Cheung, M.-S.; Day, D.S.; Gadel, S.; Gorchakov, A.A. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2011, 18, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; McCormack, A.L.; Schieltz, D.; Carmack, E.; Link, A. Direct analysis of protein mixtures by tandem mass spectrometry. J. Protein Chem. 1997, 16, 495–497. [Google Scholar] [CrossRef]
- Rabilloud, T. Paleoproteomics explained to youngsters: How did the wedding of two-dimensional electrophoresis and protein sequencing spark proteomics on: Let there be light. J. Proteom. 2014, 107, 5–12. [Google Scholar] [CrossRef]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. High Resolution Quantitative Proteomics of HeLa Cells Protein Species Using Stable Isotope Labeling with Amino Acids in Cell Culture(SILAC), Two-Dimensional Gel Electrophoresis(2DE) and Nano-Liquid Chromatography Coupled to an LTQ-Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2013, 12, 29–538. [Google Scholar]
- Han, Y.; Lu, C.; Zhang, K.; Tian, S.; Fan, E.; Chen, L.; He, X.; Zhang, Y. Quantitative characterization of histone post-translational modifications using a stable isotope dimethyl labeling strategy. Anal. Methods 2015, 7, 3779–3785. [Google Scholar] [CrossRef]
- Tian, S.; Zheng, S.; Han, Y.; Guo, Z.; Zhai, G.; Bai, X.; He, X.; Fan, E.; Zhang, Y.; Zhang, K. Maleic anhydride labeling-based approach for quantitative proteomics and successive derivatization of peptides. Anal. Chem. 2017, 89, 8259–8265. [Google Scholar] [CrossRef]
- Garcia, B.A.; Mollah, S.; Ueberheide, B.M.; Busby, S.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933–938. [Google Scholar] [CrossRef]
- Sidoli, S.; Yuan, Z.-F.; Lin, S.; Karch, K.; Wang, X.; Bhanu, N.; Arnaudo, A.M.; Britton, L.-M.; Cao, X.-J.; Gonzales-Cope, M.; et al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics 2015, 15, 1459–1469. [Google Scholar] [CrossRef]
- Wysocki, V.H.; Tsaprailis, G.; Smith, L.L.; Breci, L.A.J. Mobile and localized protons: A framework for understanding peptide dissociation. J. Mass Spectrom. 2000, 35, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A. Electron-capture dissociation tandem mass spectrometry. Curr. Opin. Biotechnol. 2004, 15, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Syka, J.E.P.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533. [Google Scholar] [CrossRef]
- Cristobal, A.; Marino, F.; Post, H.; van den Toorn, H.W.P.; Mohammed, S.; Heck, A.J. Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem. 2017, 89, 3318–3325. [Google Scholar] [CrossRef]
- Taouatas, N.; Drugan, M.M.; Heck, A.J.R.; Mohammed, S. Straightforward ladder sequencing of peptides using a Lys-N metalloendopeptidase. Nat. Methods 2008, 5, 405–407. [Google Scholar] [CrossRef]
- Hohmann, L.; Sherwood, C.; Eastham, A.; Peterson, A.; Eng, J.K.; Eddes, J.S.; Shteynberg, D.; Martin, D.B. Proteomic analyses using Grifola frondose metalloendoprotease Lys-N. J. Proteome Res. 2009, 8, 1415–1422. [Google Scholar] [CrossRef]
- Swaney, D.L.; Wenger, C.D.; Coon, J.J. Value of using multiple proteases for largescale mass spectrometry-based proteomics. J. Proteome Res. 2010, 9, 1323–1329. [Google Scholar] [CrossRef]
- Sidoli, S.; Schwämmle, V.; Ruminowicz, C.; Hansen, T.A.; Wu, X.; Helin, K.; Jensen, O.N. Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 2014, 14, 2200–2211. [Google Scholar] [CrossRef]
- Taverna, S.D.; Ueberheide, B.M.; Liu, Y.; Tackett, A.J.; Diaz, R.L.; Shabanowitz, J.; Chait, B.T.; Hunt, D.F.; Allis, C.D. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc. Natl. Acad. Sci. USA 2007, 104, 2086–2091. [Google Scholar] [CrossRef] [PubMed]
- Forbes, A.J.; Mazur, M.T.; Patel, H.M.; Walsh, C.T.; Kelleher, N.L. Toward efficient analysis of >70 kDa proteins with 100% sequence coverage. Proteomics 2001, 1, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-L.; Kim, J.; Hancock, W.S.; Karger, B. Extended Range Proteomic Analysis (ERPA): A new and sensitive LC-MS platform for high sequence coverage of complex proteins with extensive post-translational modifications-comprehensive analysis of betacasein and epidermal growth factor receptor (EGFR). J. Proteome Res. 2005, 4, 1155–1170. [Google Scholar] [CrossRef] [PubMed]
- Boyne, M.T.; Garcia, B.A.; Li, M.; Zamdborg, L.; Wenger, C.D.; Babai, S.; Kelleher, N.L. Tandem mass spectrometry with ultrahigh mass accuracy clarifies peptide identification by database retrieval. J. Proteome Res. 2009, 8, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.; Lohnes, K.; Wynne, C.; Wang, Y.; Edwards, N.; Fenselau, C. High-throughput middle-down analysis using an orbitrap. J. Proteome Res. 2010, 9, 3886–3890. [Google Scholar] [CrossRef] [PubMed]
- Mclean, J.A.; Ruotolo, B.T.; Gillig, K.J.; Russel, D.H. Ion mobility–mass spectrometry: A new paradigm for proteomics. Int. J. Mass Spectrom. 2005, 240, 301–315. [Google Scholar] [CrossRef]
- Shaibani, A.; Haghpazir, N. Application of ion mobility spectrometry for the determination of tramadol in biological samples. J. Food Drug Anal. 2014, 22, 500–504. [Google Scholar] [CrossRef]
- Morrison, K.A.; Clowers, B.H. Contemporary glycomic approaches using ion mobility–mass spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 119–129. [Google Scholar] [CrossRef]
- Ross, D.H.; Xu, L. Determination of drugs and drug metabolites by ion mobility-mass spectrometry. Anal. Chim. Acta 2021, 1154, 338270. [Google Scholar] [CrossRef]
- Ding, L.; Brancia, F.L. Electron capture dissociation in a digital ion trap mass spectrometer. Anal. Chem. 2006, 78, 1995–2000. [Google Scholar] [CrossRef]
- Zubarev, R.A.; Horn, D.M.; Fridriksson, E.K.; Kelleher, N.L.; Kruger, N.A.; Lewis, M.A.; Carpenter, B.K.; McLafferty, F.W. Electron capture dissociation for structural characterization of multiply charged proteincations. Anal. Chem. 2000, 72, 563. [Google Scholar] [CrossRef]
- Madsen, J.A.; Boutz, D.R.; Brodbelt, J.S. Ultrafast ultraviolet photodissociation at 193 nm and its applicability to proteomic workflows. J. Proteome Res. 2010, 9, 4205–4214. [Google Scholar] [CrossRef]
- Brodbelt, J.S.; Morrison, L.J.; Santos, I. Ultraviolet photodissociation mass spectrometry for analysis of biological molecules. Chem. Rev. 2020, 120, 3328–3380. [Google Scholar] [CrossRef]
- Laskin, J.; Futrell, J.H. Activation of large lons in FT-ICR massspectrometry. Mass Spectrom. Rev. 2005, 24, 135–167. [Google Scholar] [CrossRef]
- Voinov, V.G.; Hoffman, P.D.; Bennett, S.E.; Beckman, J.S.; Barofsky, D.F. Electron capture dissociation of sodium-adducted peptides on a modified quadrupole/time-of-flight mass spectrometer. J. Am. Soc. Mass Spectrom. 2015, 26, 2096–2104. [Google Scholar] [CrossRef]
- Shaw, J.B.; Malhan, N.; Vasilev, Y.V.; Lopez, N.I.; Makarov, A.; Beckman, J.S.; Voinov, V.G. Sequencing grade tandem mass spectrometry for top–down proteomics using hybrid electron capture dissociation methods in a benchtop Orbitrap mass spectrometer. Anal. Chem. 2018, 90, 10819–10827. [Google Scholar] [CrossRef]
- Voinov, V.G.; Beckman, J.S.; Deinzer, M.L.; Barofsky, D.F. Electron-capture dissociation(ECD), collision-induced dissociation(CID) and ECD/CID in a linear radio-frequency-free magnetic cell. Rapid Commun. Mass Spectrom. 2009, 23, 3028. [Google Scholar] [CrossRef]
- Thompson, M.S.; Cui, W.; Reilly, J.P. Fragmentation of singly charged peptide ions by photodissociation at λ = 157nm. Angew. Chem. Int. Ed. 2004, 43, 4791–4794. [Google Scholar]
- Cannon, J.R.; Cammarata, M.B.; Robotham, S.A.; Cotham, V.C.; Shaw, J.B.; Fellers, R.T.; Early, B.P.; Thomas, P.M.; Kelleher, N.L.; Brodbelt, J.S. Ultraviolet photodissociation for characterization of whole proteins on a chromatographic time scale. Anal. Chem. 2014, 86, 2185–2192. [Google Scholar] [CrossRef]
- Shaw, J.B.; Li, W.; Holden, D.D.; Zhang, Y.; Griep-Raming, J.; Fellers, R.T.; Early, B.P.; Thomas, P.M.; Kelleher, N.L.; Brodbelt, J.S. Complete protein characterization using top-down mass spectrometry and ultraviolet photodissociation. J. Am. Chem. Soc. 2013, 135, 12646–12651. [Google Scholar] [CrossRef]
- Smyrnakis, A.; Levin, N.; Kosmopoulou, M.; Fort, K.; Makarov, A.; Papanastasiou, D.; Mohammed, S. Characterization of an Omnitrap-Orbitrap Platform Equipped with Infrared Multiphoton Dissociation, Ultraviolet Photodissociation, and Electron Capture Dissociation for the Analysis of Peptides and Proteins. Anal. Chem. 2023, 95, 32. [Google Scholar] [CrossRef]
- Papanastasiou, D.; Kounadis, D.; Lekkas, A.; Orfanopoulos, I.; Mpozatzidis, A.; Smyrnakis, A.; Panagiotopoulos, E.; Kosmopoulou, M.; Reinhardt-Szyba, M.; Fort, K.; et al. The Omnitrap platform: A versatile segmented linear ion trap for multidimensional multiple-stage tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2022, 33, 1990–2007. [Google Scholar] [CrossRef]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Greger, V.; Passarge, E.; Höpping, W.; Messmer, E.; Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet. 1989, 83, 155–158. [Google Scholar] [CrossRef]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef]
- Yozbaki, M.; Jabre, I.; Syed, N.H.; Wilson, C.M. Targeting DNA methyltransferases in non-small-cell lung cancer. Semin. Cancer Biol. 2022, 83, 77–87. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 2009, 10, 192–206. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Crofton, J. Chemotherapy of pulmonary tuberculosis. Br. Med. J. 1959, 1, 1610–1614. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, G.; Brusamolino, E.; Valagussa, P.; Rossi, A.; Brugnatelli, L.; Brambilla, C.; De Lena, M.; Tancini, G.; Bajetta, E.; Musumeci, R.; et al. Combination chemotherapy as an adjuvant treatment in operable breast cancer. N. Engl. J. Med. 1976, 294, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Bosl, G.J.; Gluckman, R.; Geller, N.L.; Golbey, R.B.; Whitmore, W.F., Jr.; Herr, H.; Sogani, P.; Morse, M.; Martini, N.; Bains, M. VAB6: An effective chemotherapy regimen for patients with germ-cell tumors. J. Clin. Oncol. 1986, 4, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ge, M.; Qiao, Z.; Kong, Y.; Liang, H.; Sun, Y.; Lu, H.; Xu, Z.; Liu, H. Modulating proteasome inhibitor tolerance in multiple myeloma: An alternative strategy to reverse inevitable resistance. Br. J. Cancer 2021, 124, 770–776. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Barrio, S.; Stühmer, T.; Da-Viá, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019, 33, 447–456. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Shen, F.; Chu, S.A.; Bailey, B.; Xue, X.; Erickson, P.A.; Montrose, M.H.; Beck, W.T.; Erickson, L.C. Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J. Pharmacol. Exp. Ther. 2008, 324, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.M.; Reyes-Mugica, M.; Barquin, N.; Yasuda, T.; Tan, X.; Tomita, T. Multidrug resistance gene expression in childhood medulloblastoma: Correlation with clinical outcome and DNA ploidy in 29 patients. Pediatr. Neurosurg. 1995, 23, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Zou, L.; Zhang, T.; Jiang, L.; Ding, Y.; Yu, P.; Peng, J. Using LC–MS/MS based targeted proteomics to monitor the pattern of transporters expression the development of drug resistance. Cancer Manag. Res. 2018, 10, 2859–2870. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.; Borges, C.R.; Schaab, M.R.; Billheimer, D.; Stump, C.; Reaven, P.; Lau, S.S.; Nelson, R. Mass spectrometric immunoassay and MRM as targeted MS-based quantitative approaches in biomarker development: Potential. applications to cardiovascular disease and diabetes. Proteomics Clin. Appl. 2013, 7, 528–540. [Google Scholar] [CrossRef]
- Xue, Z.; Zeng, J.; Yin, X.; Li, Y.; Meng, B.; Zhao, Y.; Fang, X.; Gong, X.; Dai, X. Investigation on acquired palbociclib resistance by LCMS based multi-omics analysis. Front. Mol. Biosci. 2023, 10, 1116398. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Borland, M.; Kottegoda, S.; Phillips, K.S.; Allbritton, N.L. Chemical analysis of single cells. Annu. Rev. Anal. Chem. 2008, 1, 191–227. [Google Scholar] [CrossRef]
- Heinemann, M.; Zenobi, R. Single cell metabolomics. Curr. Opin. Biotechnol. 2011, 22, 26–31. [Google Scholar] [CrossRef]
- Amantonico, A.; Urban, P.L.; Zenobi, R. Analytical techniques for single-cell metabolomics: State of the art and trends. Anal. Bioanal. Chem. 2010, 398, 2493–2504. [Google Scholar] [CrossRef]
- Liu, R.; Sun, M.; Zhang, G.; Lan, Y.; Yang, Z. Towards early monitoring of chemotherapy-induced drug resistance based on single cell metabolomics: Combining single-probe mass spectrometry with machine learning. Anal. Chim. Acta 2019, 1092, 42–48. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Fruci, D.; Cho, W.C.S.; Nobili, V.; Locatelli, F.; Alisi, A. Drug Transporters and Multiple Drug Resistance in Pediatric Solid Tumors. Curr. Drug Metab. 2016, 17, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Pizer, B.L.; Clifford, S.C. The potential impact of tumour biology on improved clinical practice for medulloblastoma: Progress towards biologically driven clinical trials. Br. J. Neurosurg. 2009, 23, 364–375. [Google Scholar] [CrossRef]
- Taylor, L.; Wade, P.K.; Johnson, J.E.C.; Aldighieri, M.; Morlando, S.; Di Leva, G.; Kerr, I.D.; Coyle, B. Drug Resistance in Medulloblastoma is driven by YB-1, ABCB1 and a Seven-Gene Drug Signature. Cancers 2023, 15, 1086. [Google Scholar] [CrossRef]
- Eliseeva, I.A.; Kim, E.R.; Guryanov, S.G.; Ovchinnikov, L.P.; Lyabin, D.N. Y-boxbinding protein 1 (YB-1) and its functions. Biochemistry 2011, 76, 1402–1433. [Google Scholar]
- Wang, Y.; Chen, Y.; Geng, H.; Qi, C.; Liu, Y.; Yue, D. Overexpression of YB1 and EZH2 are associated with cancer metastasis and poor prognosis in renal cell carcinomas. Tumor Biol. 2015, 36, 7159–7166. [Google Scholar] [CrossRef]
- Wang, X.; Guo, X.B.; Shen, X.C.; Zhou, H.; Wan, D.W.; Xue, X.F.; Han, Y.; Yuan, B.; Zhou, J.; Zhao, H.; et al. Prognostic role of YB-1 expression in breast cancer: A meta analysis. Int. J. Clin. Exp. Med. 2015, 8, 1780–1791. [Google Scholar]
- Bennett, J.; Ashmawy, R.; Ramaswamy, V.; Stephens, D.; Bouffet, E.; Laperriere, N.; Taylor, M.; Shroff, M.; Bartels, U. The clinical significance of equivocal findings on spinal MRI in children with medulloblastoma. Pediatr. Blood Cancer 2017, 64, e26472. [Google Scholar] [CrossRef]
- Paine, M.R.L.; Liu, J.; Huang, D.; Ellis, S.R.; Trede, D.; Kobarg, J.H.; Heeren, R.M.A.; Fernández, F.M.; MacDonald, T.J. Three-Dimensional Mass Spectrometry Imaging Identifies Lipid Markers of Medulloblastoma Metastasis. Sci. Rep. 2019, 9, 2205. [Google Scholar] [CrossRef]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Leo, I.R.; Aswad, L.; Stahl, M.; Kunold, E.; Post, F.; Erkers, T.; Struyf, N.; Mermelekas, G.; Joshi, R.N.; Gracia-Villacampa, E.; et al. Integrative multi-omics and drug response profiling of childhood acute lymphoblastic acute leukemia cell lines. Nat. Commun. 2022, 13, 1691. [Google Scholar] [CrossRef]
- Nguyen, K.; Devidas, M.; Cheng, S.-C.; La, M.; A Raetz, E.; Carroll, W.L.; Winick, N.J.; Hunger, S.P.; Gaynon, P.S.; Loh, M.L.; et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: A Children’s Oncology Group study. Leukemia 2008, 22, 2142–2150. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Söderhäll, S.; Arvidson, J.; Forestier, E.; Montgomery, S.; Bottai, M.; Lausen, B.; Carlsen, N.; Hellebostad, M.; Lähteenmäki, P.; et al. Relapsed childhood acute lymphoblastic leukemia in the Nordic countries: Prognostic factors, treatment and outcome. Haematologica 2016, 101, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.A.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Mehrazma, M.; Madjd, Z.; Kalantari, E.; Panahi, M.; Hendi, A.; Shariftabrizi, A. Expression of stem cell markers, CD133 and CD44, in pediatric solid tumors: A study using tissue microarray. Fetal Pediatr. Pathol. 2013, 32, 192–204. [Google Scholar] [CrossRef]
- Xi, G.; Li, Y.D.; Grahovac, G.; Rajaram, V.; Wadhwani, N.; Pundy, T.; Mania-Farnell, B.; James, C.D.; Tomita, T. Targeting CD133 improves chemotherapeutic efficacy of recurrent pediatric pilocytic astrocytoma following prolonged chemotherapy. Mol. Cancer. 2017, 16, 21. [Google Scholar] [CrossRef]
- Shah, N. Dodging the bullet: Therapeutic resistance mechanisms in pediatric cancers. Cancer Drug Resist. 2019, 2, 428–446. [Google Scholar] [CrossRef]
- Romac, J.; Bouley, J.P.; Van Regenmortel, M.H. Enzyme-linked immunosorbent assay in the study of histone antigens and nucleosome structure. Anal. Biochem. 1981, 113, 366–371. [Google Scholar] [CrossRef]
- Costa, O.; Monier, J.C. Detection of antibodies to histones in human systemic lupus erythematosus and in murine lupus-like syndromes using micro-ELISA. Ann. Immunol. 1983, 134C, 365–376. [Google Scholar] [CrossRef]
- Hoofnagle, A.N.; Wener, M.H. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J. Immunol. Methods 2009, 347, 3–11. [Google Scholar] [CrossRef] [PubMed]
- García-Giménez, J.L.; Romá-Mateo, C.; Carbonell, N.; Palacios, L.; Peiró-Chova, L.; García-López, E.; García-Simón, M.; Lahuerta, R.; Gimenez-Garzó, C.; Berenguer-Pascual, E.; et al. A new mass spectrometry-based method for the quantification of histones in plasma from septic shock patients. Sci. Rep. 2017, 7, 10643. [Google Scholar] [CrossRef] [PubMed]
- Holdenrieder, S.; Stieber, P.; Bodenmüller, H.; Busch, M.; Fertig, G.; Fürst, H.; Schalhorn, A.; Schmeller, N.; Untch, M.; Seidel, D. Nucleosomes in serum of patients with benign and malignant diseases. Int. J. Cancer 2001, 95, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Yörüker, E.E.; Holdenrieder, S.; Gezer, U. Potential of circulating nucleosome associated histone modifications in cancer. Transl. Cancer Res. 2017, 7, S185–S191. [Google Scholar] [CrossRef]
- McAnena, P.; Brown, J.A.L.; Kerin, M.J. Circulating nucleosomes and nucleosome modifications as biomarkers in cancer. Cancers 2017, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Stoetzer, O.J.; Fersching, D.M.I.; Salat, C.; Steinkohl, O.; Gabka, C.J.; Hamann, U.; Braun, M.; Feller, A.M.; Heinemann, V.; Siegele, B.; et al. Prediction of response to neoadjuvant chemotherapy in breast cancer patients by circulating apoptotic biomarkers nucleosomes, DNAse, cytokeratin-18 fragments and survivin. Cancer Lett. 2013, 336, 140–148. [Google Scholar] [CrossRef]
- Van den Ackerveken, P.; Lobbens, A.; Turatsinze, J.-V.; Solis-Mezarino, V.; Völker-Albert, M.; Imhof, A.; Herzog, M. A novel proteomics approach to epigenetic profiling of circulating nucleosomes. Sci. Rep. 2021, 11, 7256. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agostini, M.; Traldi, P.; Hamdan, M. Mass Spectrometry-Based Proteomics: Analyses Related to Drug-Resistance and Disease Biomarkers. Medicina 2023, 59, 1722. https://doi.org/10.3390/medicina59101722
Agostini M, Traldi P, Hamdan M. Mass Spectrometry-Based Proteomics: Analyses Related to Drug-Resistance and Disease Biomarkers. Medicina. 2023; 59(10):1722. https://doi.org/10.3390/medicina59101722
Chicago/Turabian StyleAgostini, Marco, Pietro Traldi, and Mahmoud Hamdan. 2023. "Mass Spectrometry-Based Proteomics: Analyses Related to Drug-Resistance and Disease Biomarkers" Medicina 59, no. 10: 1722. https://doi.org/10.3390/medicina59101722