

CDHR1-Related Cone–Rod Dystrophy: Clinical Characteristics, Imaging Findings, and Genetic Test Results—A Case Report

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
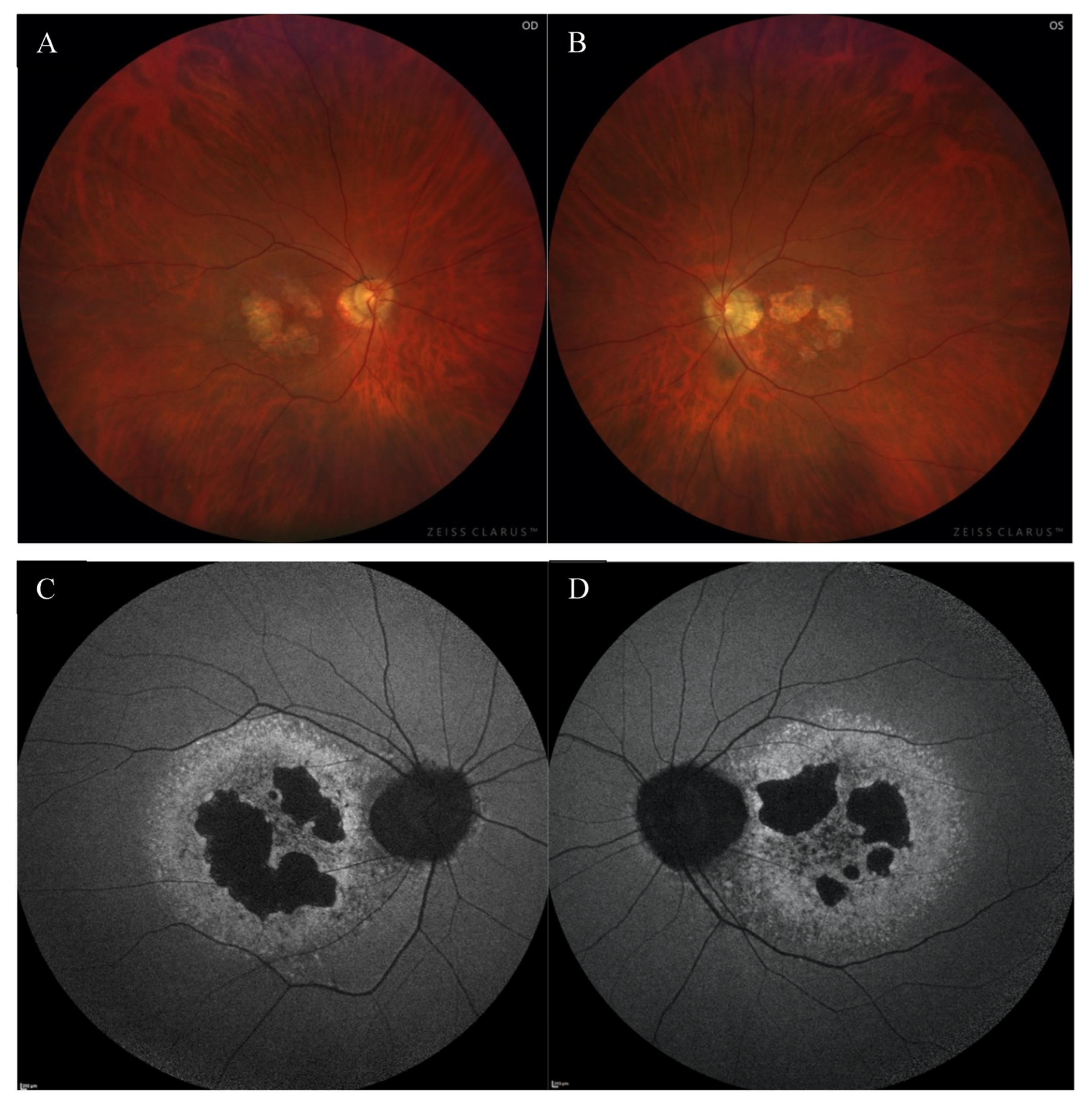
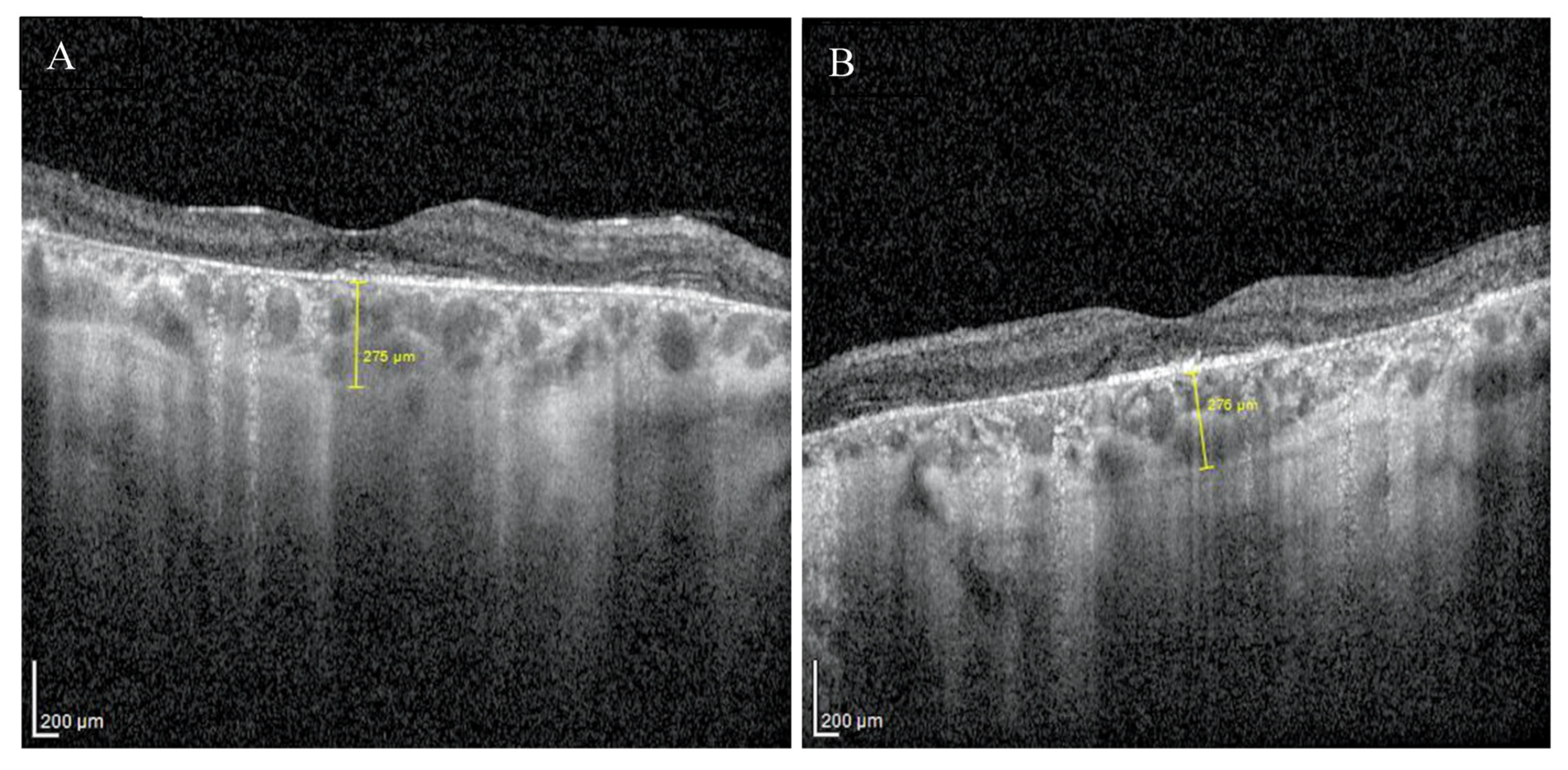
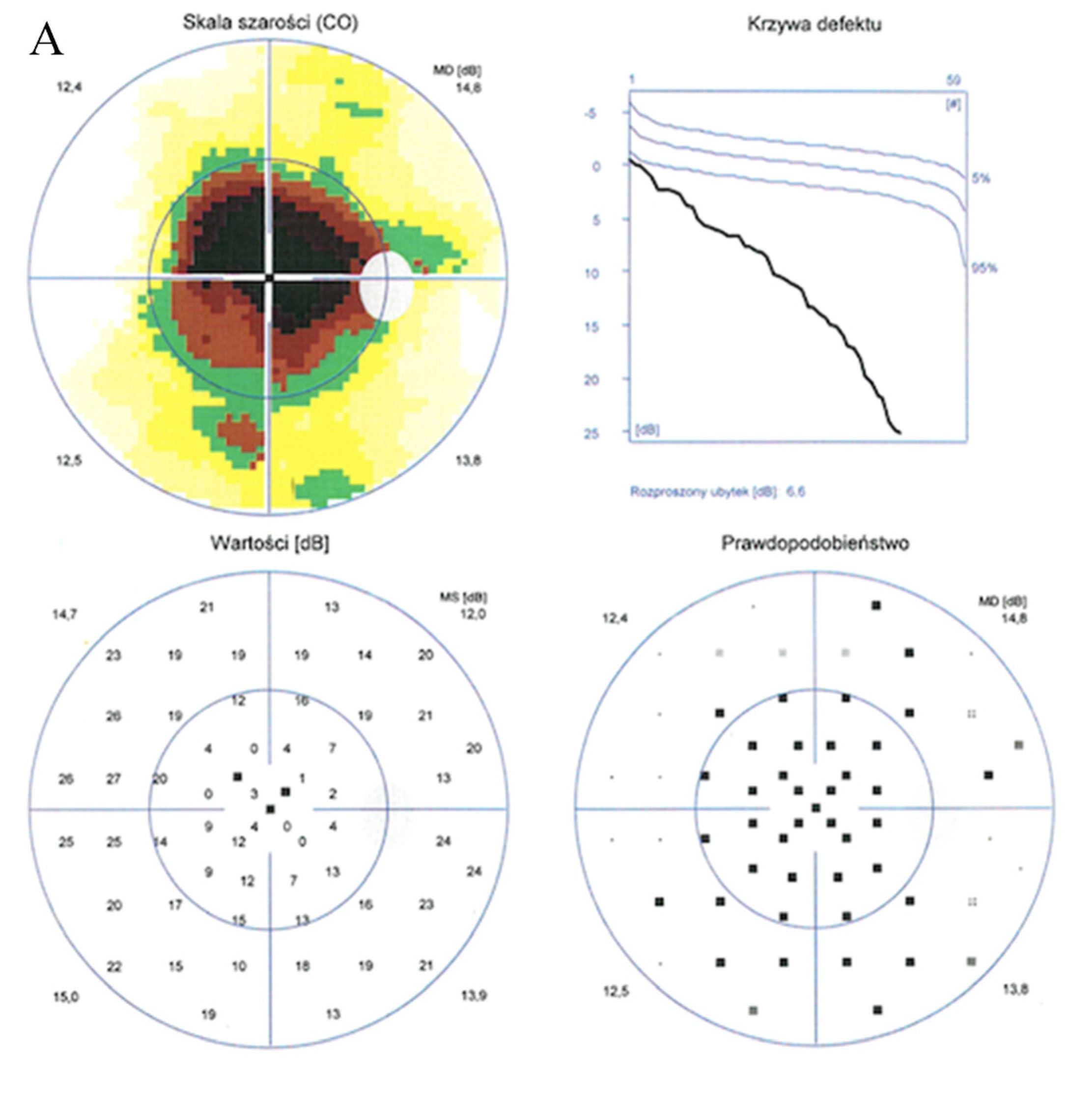
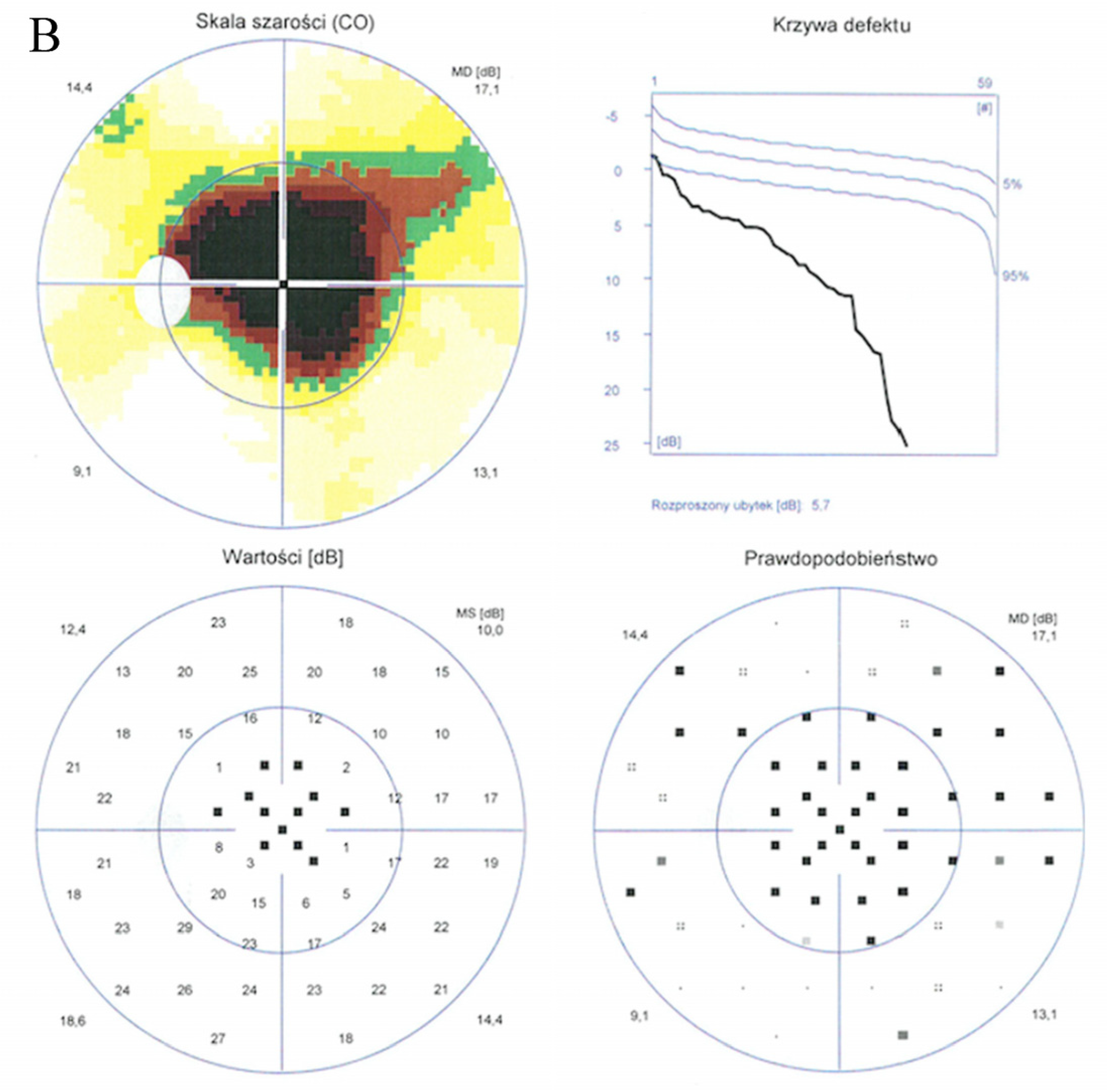
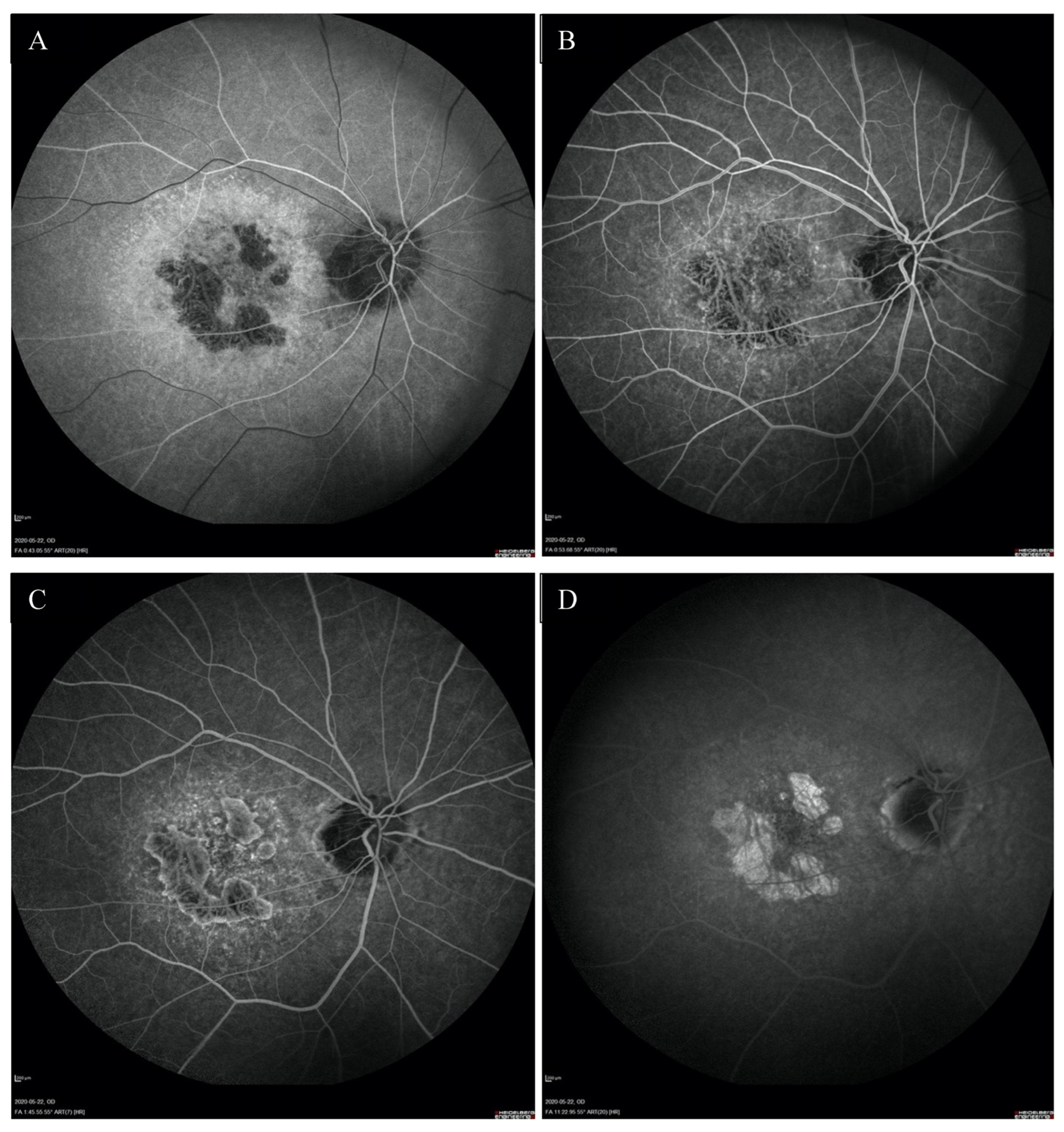
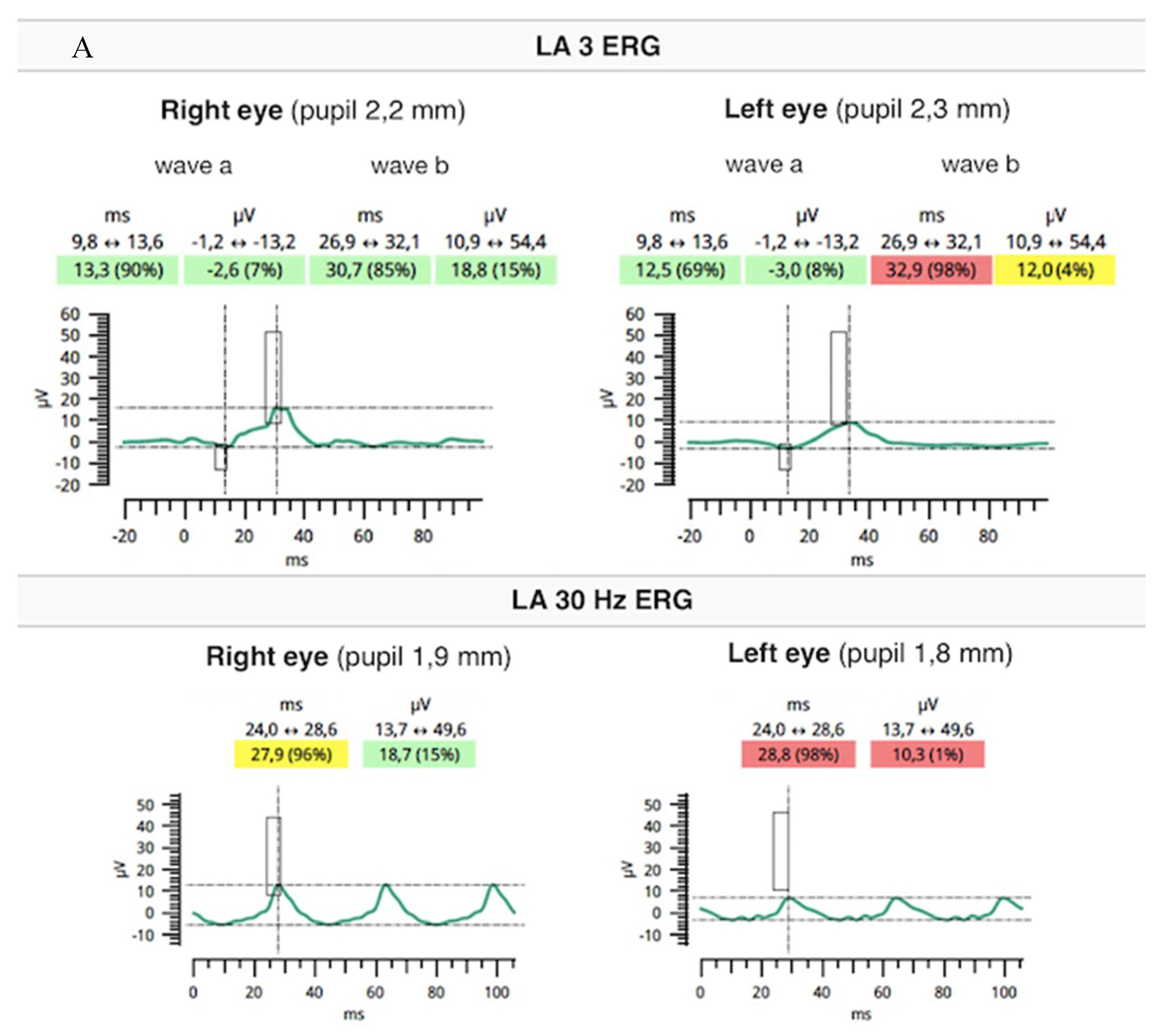
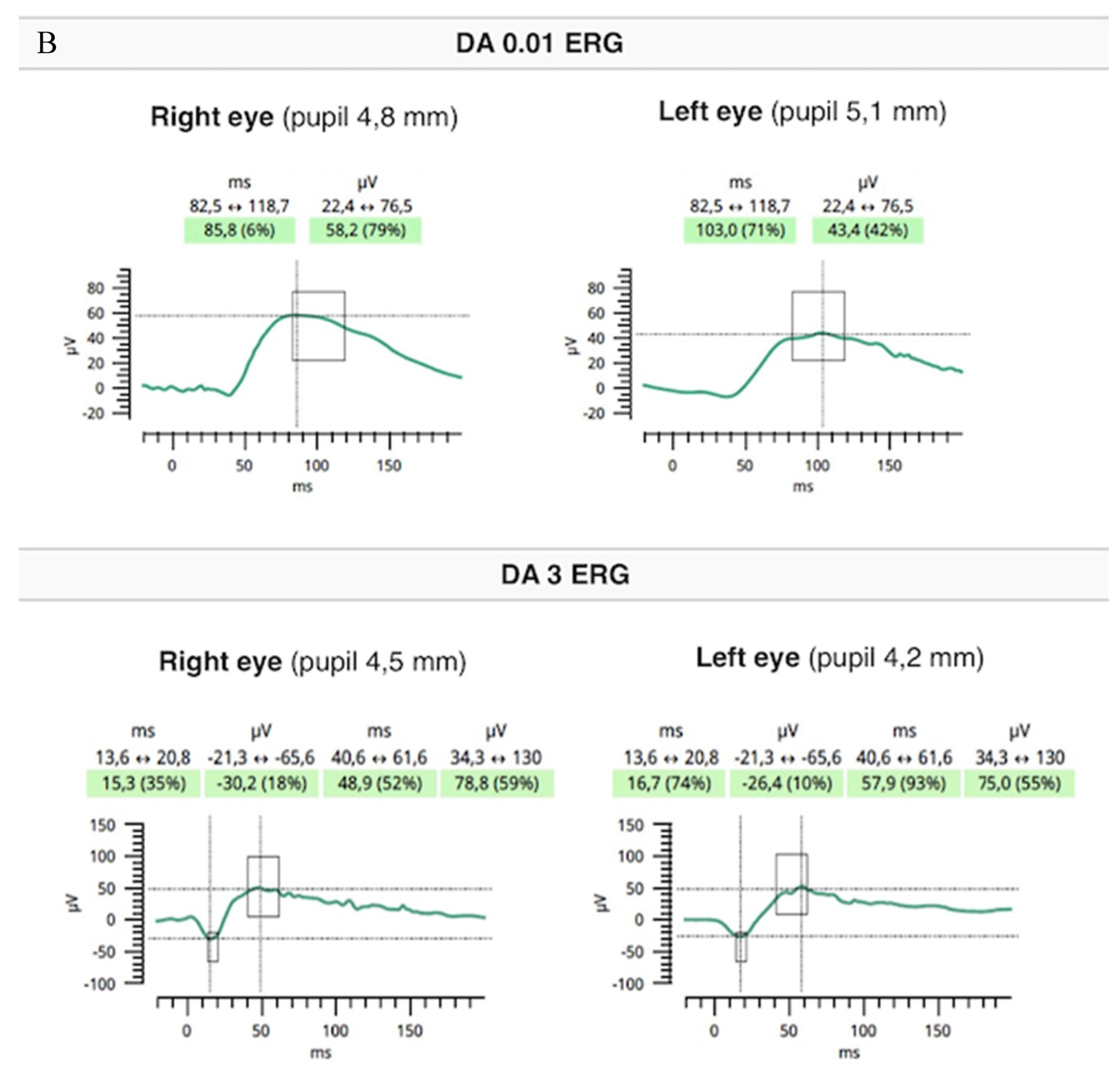
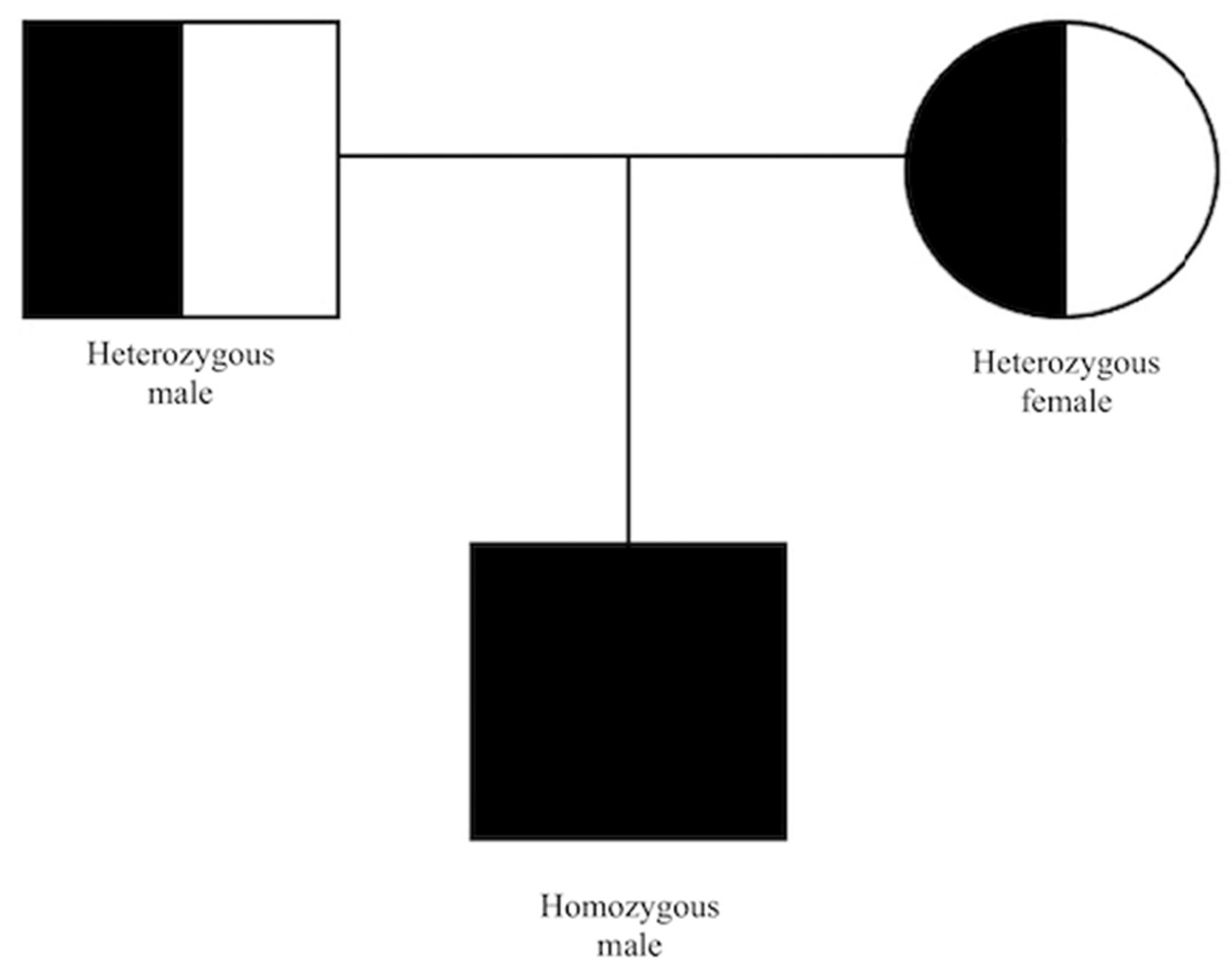
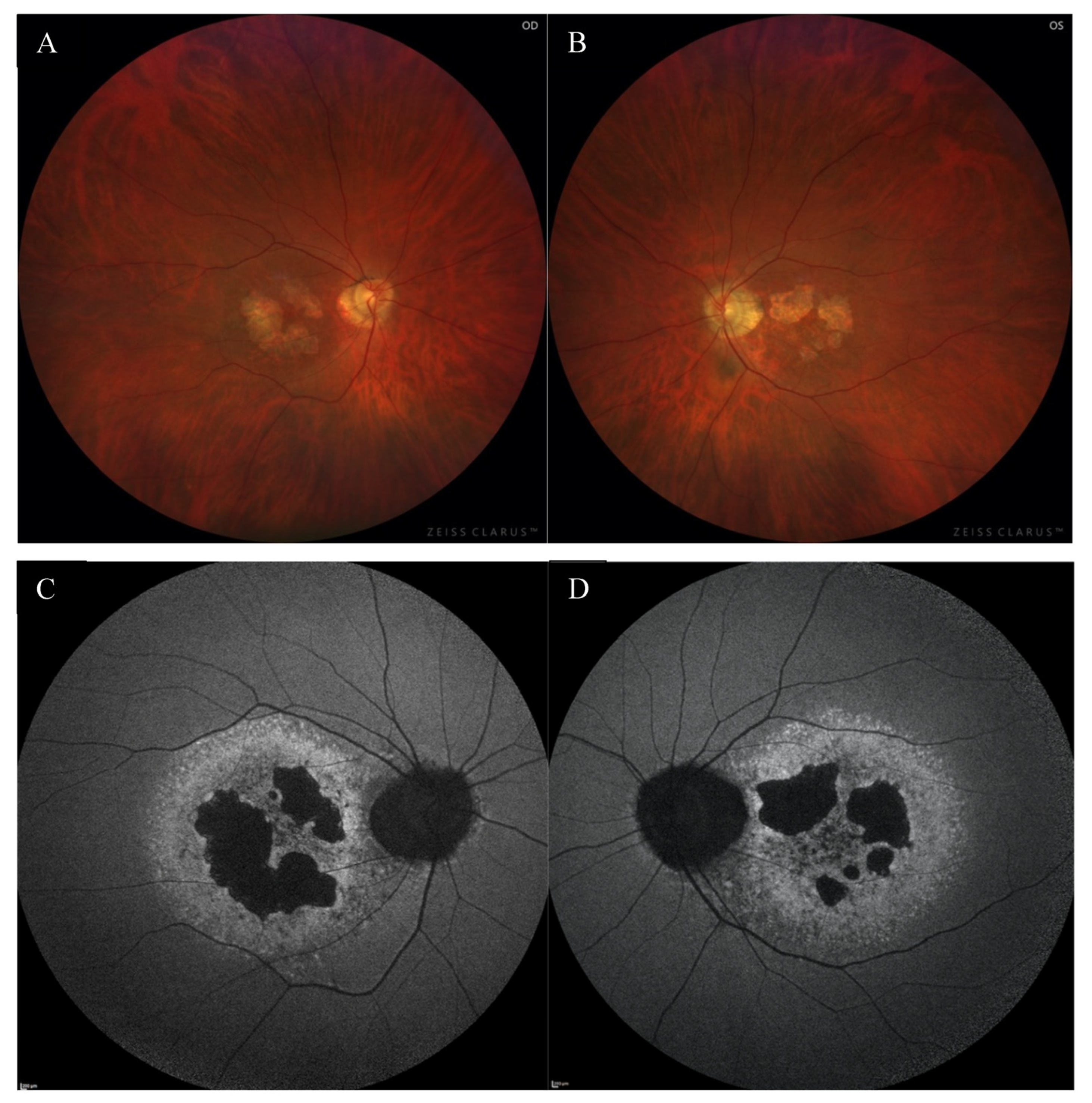
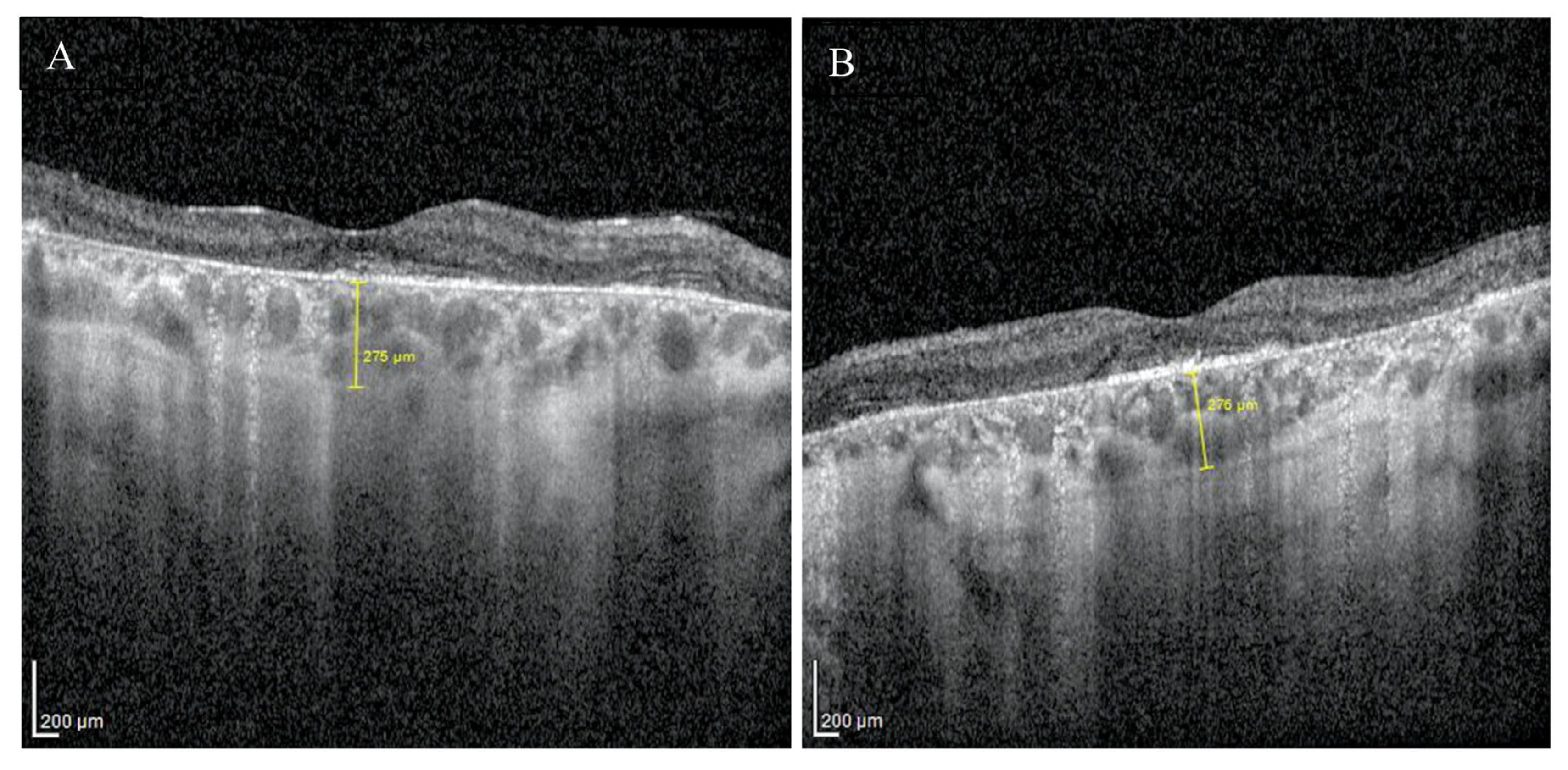
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aboshiha, J.; Dubis, A.M.; Carroll, J.; Hardcastle, A.J.; Michaelides, M. The cone dysfunction syndromes. Br. J. Ophthalmol. 2016, 100, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Michaelides, M.; Hardcastle, A.J.; Hunt, D.M.; Moore, A.T. Progressive cone and cone-rod dystrophies: Phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 2006, 51, 232–258. [Google Scholar] [CrossRef]
- Hamel, C.P. Cone rod dystrophies. Orphanet. J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Hamel, C.P.; Griffoin, J.M.; Bazalgette, C.; Lasquellec, L.; Duval, P.A.; Bareil, C.; Beaufrère, L.; Bonnet, S.; Eliaou, C.; Marlhens, F.; et al. Génétique moléculaire des rétinopathies pigmentaires: Identification de mutations des gènes CHM, RDS, RHO, RPE65, USH2A et XLRS1. J. Fr. Ophtalmol. 2000, 23, 985–995. [Google Scholar]
- Tsang, S.H.; Sharma, T. Progressive Cone Dystrophy and Cone-Rod Dystrophy (XL, AD, and AR). In Atlas of Inherited Retinal Diseases. Advances in Experimental Medicine and Biology; Tsang, S., Sharma, T., Eds.; Springer: Cham, Switzerland, 2018; Volume 1085. [Google Scholar] [CrossRef]
- Stingl, K.; Mayer, A.K.; Llavona, P.; Mulahasanovic, L.; Rudolph, G.; Jacobson, S.G.; Zrenner, E.; Kohl, S.; Wissinger, B.; Weisschuh, N. CDHR1 mutations in retinal dystrophies. Sci. Rep. 2017, 7, 6992. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Fishman, G.A.; Stone, E.M.; Eliason, D.A.; Taylor, C.M.; Lindeman, M.; Derlacki, D.J. ABCA4 gene sequence variations in patients with autosomal recessive cone-rod dystrophy. Arch. Ophthalmol. 2003, 121, 851–855. [Google Scholar] [CrossRef] [Green Version]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef]
- Kellner, U.; Sadowski, B.; Zrenner, E.; Foerster, M.H. Selective cone dystrophy with protan genotype. Investig. Ophthalmol. Vis. Sci. 1995, 36, 2381–2387. [Google Scholar]
- Went, L.N.; van Schooneveld, M.J.; Oosterhuis, J.A. Late onset dominant cone dystrophy with early blue cone involvement. J. Med. Genet. 1992, 29, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Traboulsi, E.I. (Ed.) Cone Dysfunction Syndromes, Cone Dystrophies, and Cone-Rod Degenerations. In Genetic Diseases of the Eye, 2nd ed.; Oxford Monographs on Medical Genetics, Oxford Academic: New York, NY, USA, 2012; pp. 410–420. [Google Scholar] [CrossRef]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [Green Version]
- Robson, A.G.; Michaelides, M.; Luong, V.A.; Holder, G.E.; Bird, A.C.; Webster, A.R.; Moore, A.T.; Fitzke, F.W. Functional correlates of fundus autofluorescence abnormalities in patients with RPGR or RIMS1 mutations causing cone or cone rod dystrophy. Br. J. Ophthalmol. 2008, 92, 95–102. [Google Scholar] [CrossRef]
- Charbel Issa, P.; Gliem, M.; Yusuf, I.H.; Birtel, J.; Müller, P.L.; Mangold, E.; Downes, S.M.; MacLaren, R.E.; Betz, C.; Bolz, H.J. A Specific Macula-Predominant Retinal Phenotype Is Associated with the CDHR1 Variant c.783G>A, a Silent Mutation Leading to In-Frame Exon Skipping. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3388–3397. [Google Scholar] [CrossRef] [Green Version]
- Ayyildiz, O.; Ozge, G.; Kucukevcilioglu, M.; Ozgonul, C.; Mumcuoglu, T.; Durukan, A.H.; Mutlu, F.M. Is there a relationship between outer retinal destruction and choroidal changes in cone dystrophy? Arq. Bras. Oftalmol. 2016, 79, 315–318. [Google Scholar] [CrossRef] [Green Version]
- Sabbaghi, H.; Ahmadieh, H.; Jalili, J.; Behnaz, N.; Fakhri, M.; Suri, F.; Kheiri, B.; Rajabpour, M.; Entezari, M.; Daftarian, N. Choroidal Thickness in Different Types of Inherited Retinal Dystrophies. J. Ophthalmic Vis. Res. 2020, 15, 351–361. [Google Scholar] [CrossRef]
- Bessette, A.P.; DeBenedictis, M.J.; Traboulsi, E.I. Clinical characteristics of recessive retinal degeneration due to mutations in the CDHR1 gene and a review of the literature. Ophthalmic Genet. 2018, 39, 51–55. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, H.; Dizhoor, A.M.; Boye, S.E.; Hauswirth, W.W.; Frederick, J.M.; Baehr, W. Long-term RNA interference gene therapy in a dominant retinitis pigmentosa mouse model. Proc. Natl. Acad. Sci. USA 2011, 108, 18476–18481. [Google Scholar] [CrossRef] [Green Version]
- Sarra, G.M.; Stephens, C.; de Alwis, M.; Bainbridge, J.W.; Smith, A.J.; Thrasher, A.J.; Ali, R.R. Gene replacement therapy in the retinal degeneration slow (rds) mouse: The effect on retinal degeneration following partial transduction of the retina. Hum. Mol. Genet. 2001, 10, 2353–2361. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Conley, S.M.; Makkia, R.S.; Cooper, M.J.; Naash, M.I. DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J. Clin. Investig. 2012, 122, 3221–3226. [Google Scholar] [CrossRef]
- Beltran, W.A.; Cideciyan, A.V.; Lewin, A.S.; Hauswirth, W.W.; Jacobson, S.G.; Aguirre, G.D. Gene augmentation for X-linked retinitis pigmentosa caused by mutations in RPGR. Cold Spring Harb. Perspect. Med. 2014, 5, a017392. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobolewska, M.; Świerczyńska, M.; Dorecka, M.; Wyględowska-Promieńska, D.; Krawczyński, M.R.; Mrukwa-Kominek, E. CDHR1-Related Cone–Rod Dystrophy: Clinical Characteristics, Imaging Findings, and Genetic Test Results—A Case Report. Medicina 2023, 59, 399. https://doi.org/10.3390/medicina59020399
Sobolewska M, Świerczyńska M, Dorecka M, Wyględowska-Promieńska D, Krawczyński MR, Mrukwa-Kominek E. CDHR1-Related Cone–Rod Dystrophy: Clinical Characteristics, Imaging Findings, and Genetic Test Results—A Case Report. Medicina. 2023; 59(2):399. https://doi.org/10.3390/medicina59020399
Chicago/Turabian StyleSobolewska, Małgorzata, Marta Świerczyńska, Mariola Dorecka, Dorota Wyględowska-Promieńska, Maciej R. Krawczyński, and Ewa Mrukwa-Kominek. 2023. "CDHR1-Related Cone–Rod Dystrophy: Clinical Characteristics, Imaging Findings, and Genetic Test Results—A Case Report" Medicina 59, no. 2: 399. https://doi.org/10.3390/medicina59020399
APA StyleSobolewska, M., Świerczyńska, M., Dorecka, M., Wyględowska-Promieńska, D., Krawczyński, M. R., & Mrukwa-Kominek, E. (2023). CDHR1-Related Cone–Rod Dystrophy: Clinical Characteristics, Imaging Findings, and Genetic Test Results—A Case Report. Medicina, 59(2), 399. https://doi.org/10.3390/medicina59020399