
DLC1 as Druggable Target for Specific Subsets of Gastric Cancer: An RNA-seq-Based Study


Abstract
:1. Introduction
2. Materials and Method
2.1. Datasets
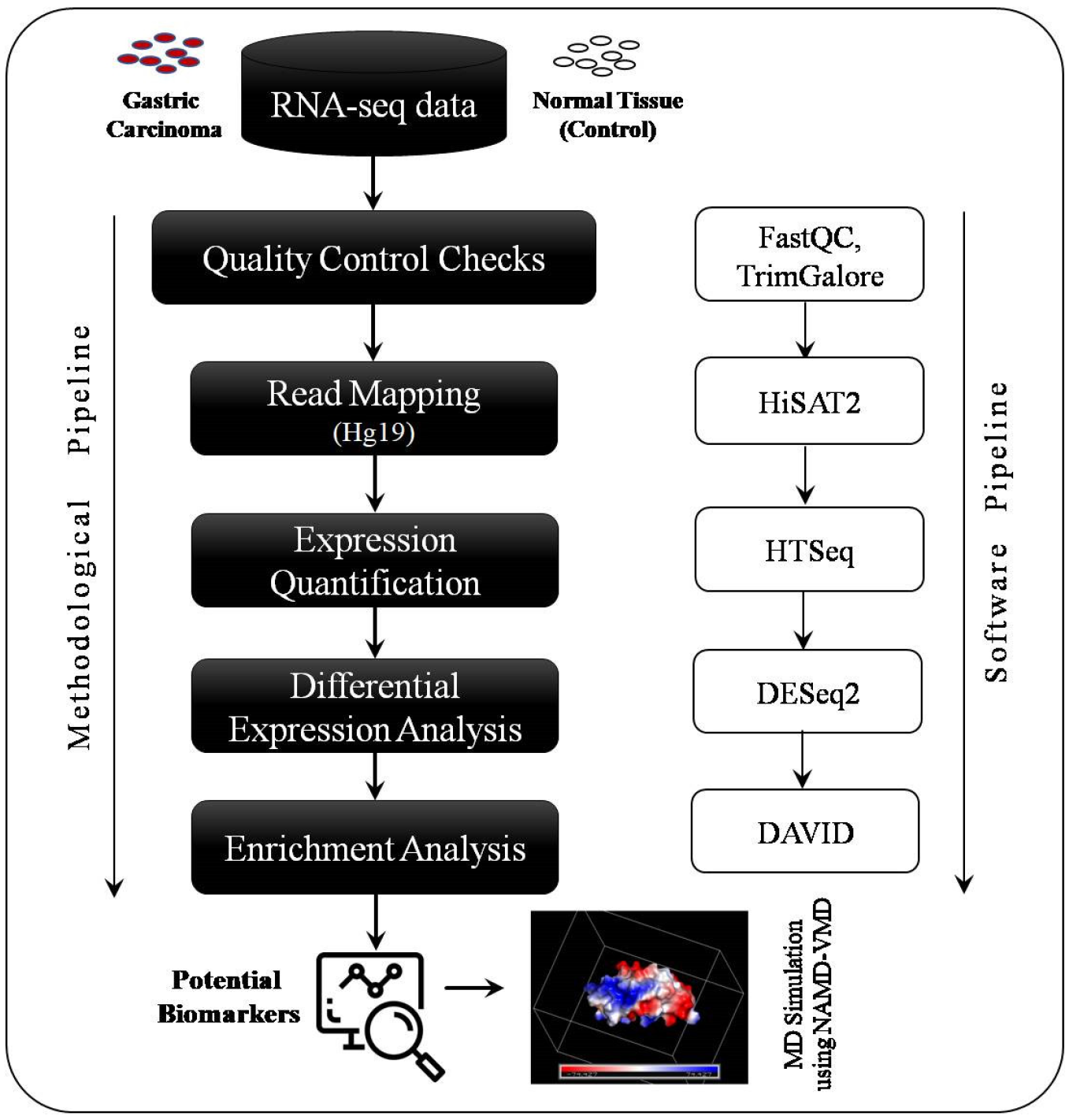
2.2. Data Processing Pipeline for DEGs’ Identification
2.3. GO and KEGG Pathway Enrichment Analysis
2.4. Molecular Dynamics Simulation
3. Results
3.1. Read Mapping
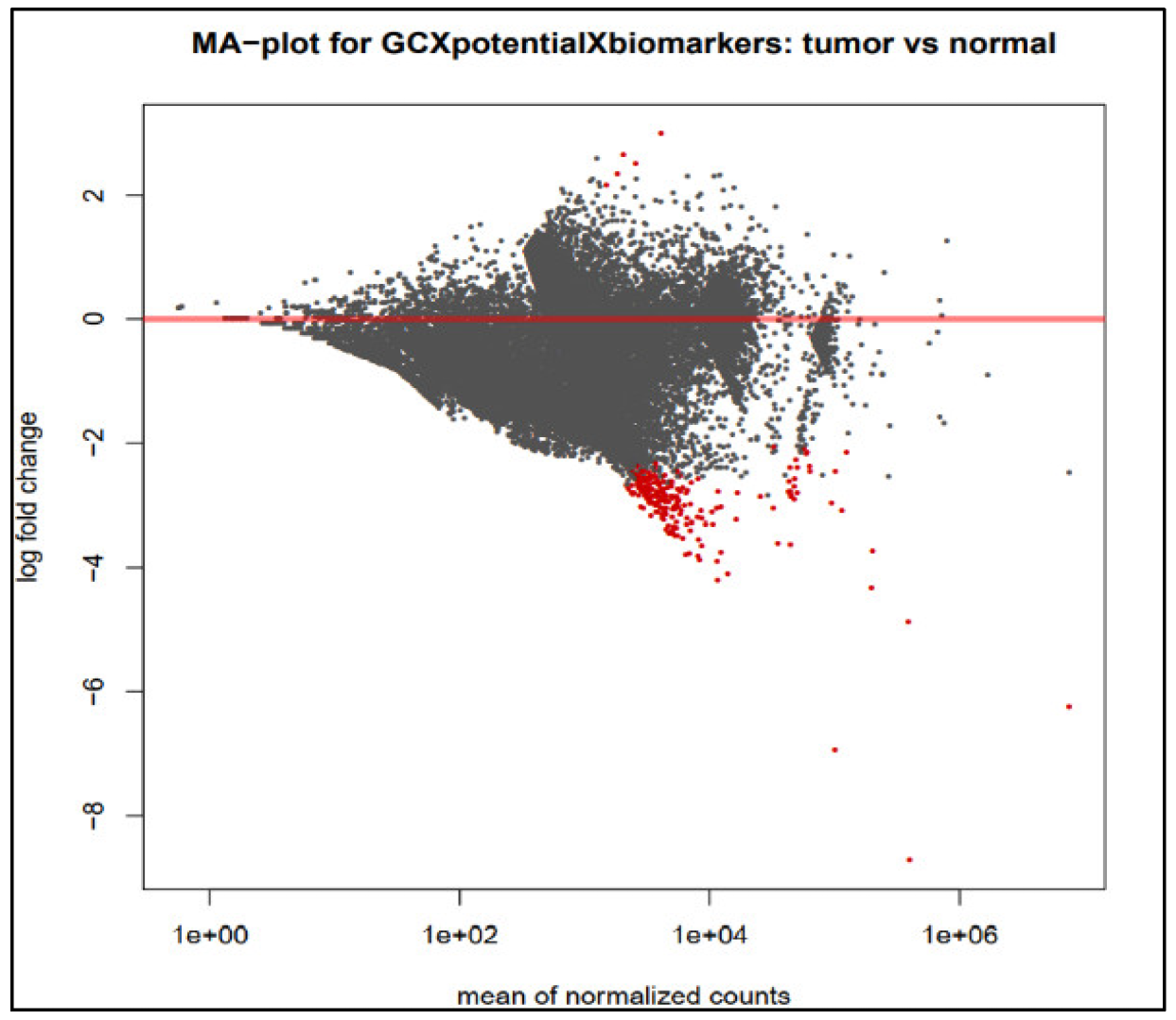
3.2. Identification of Differentially Expressed Genes
3.3. GO and KEGG Pathway Enrichment Analysis
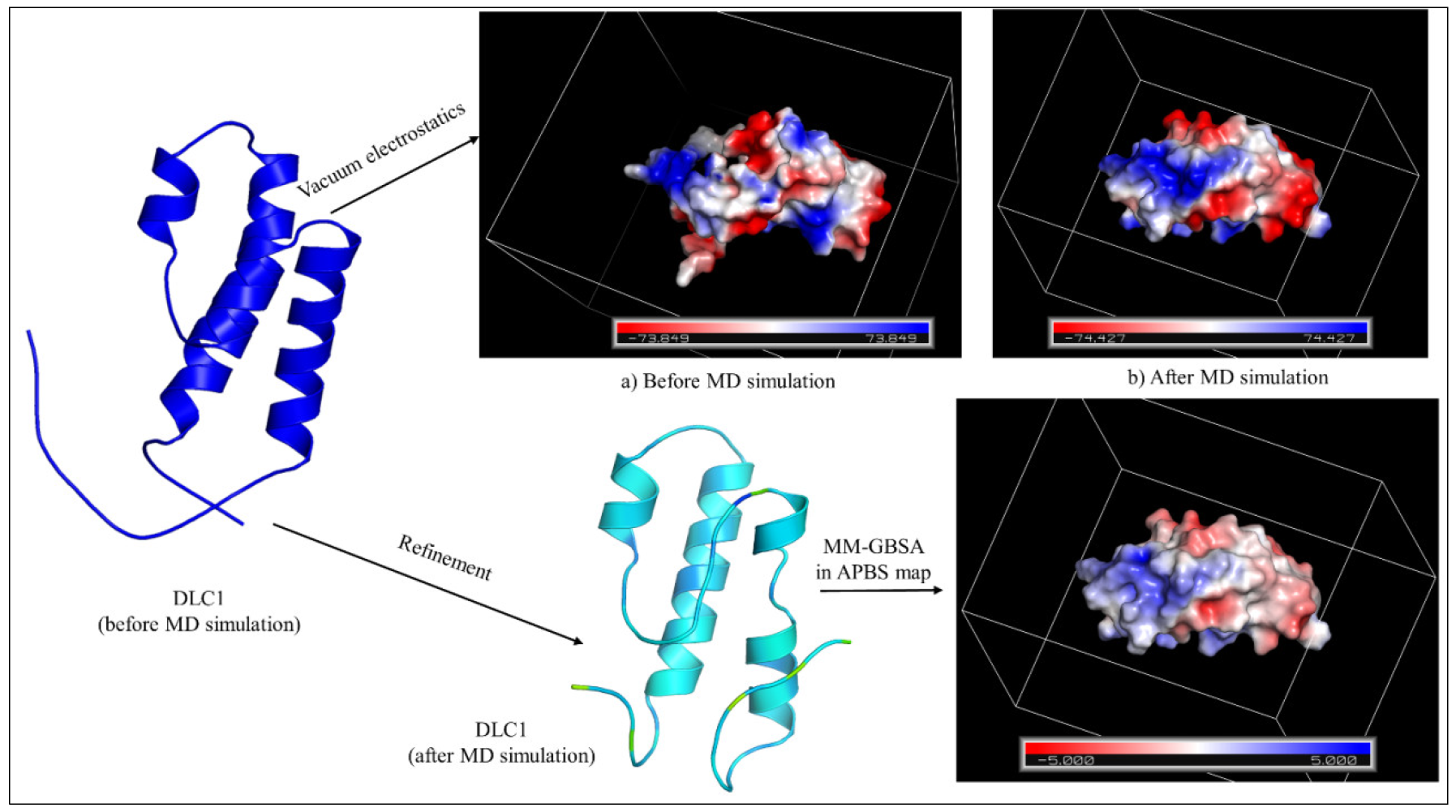
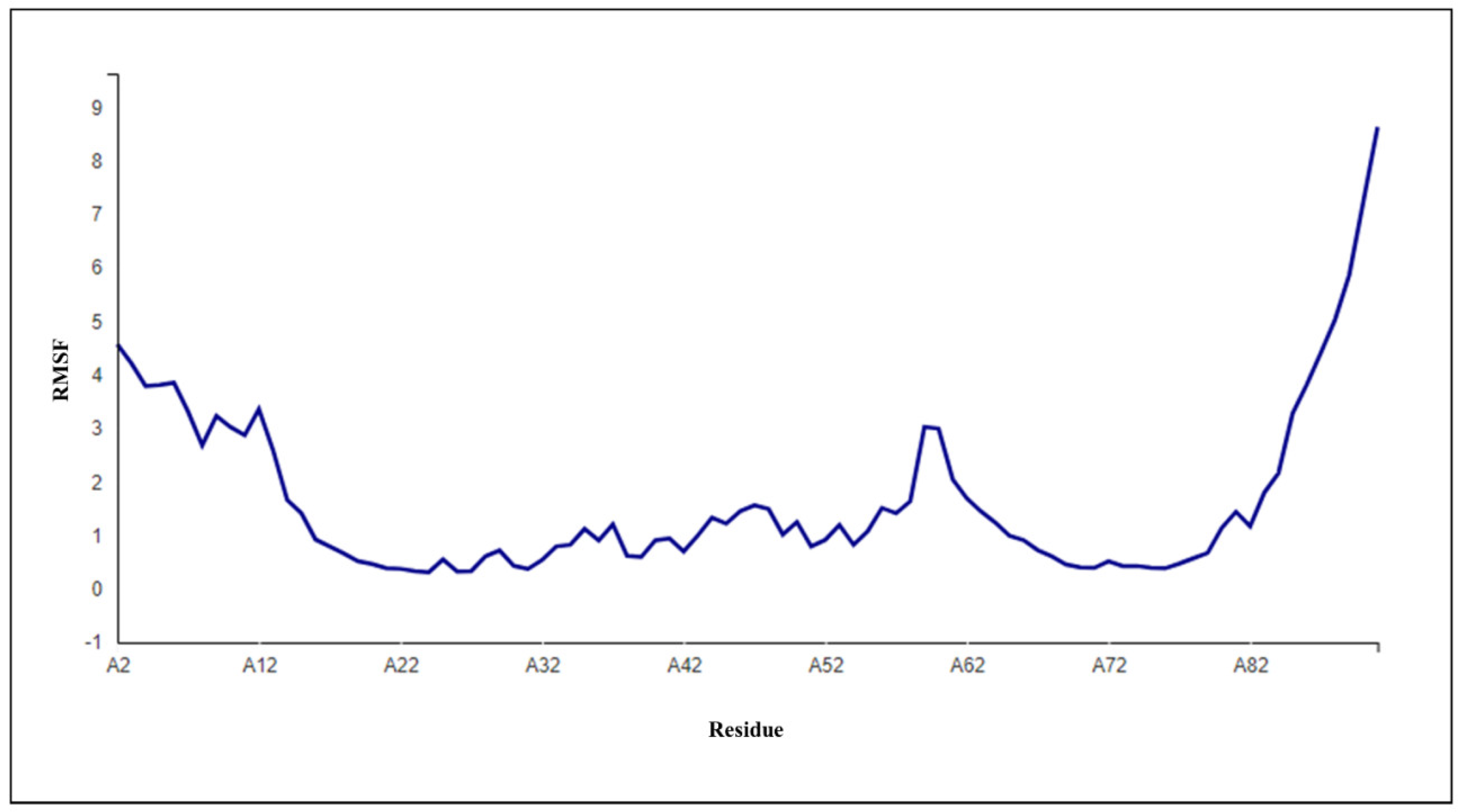
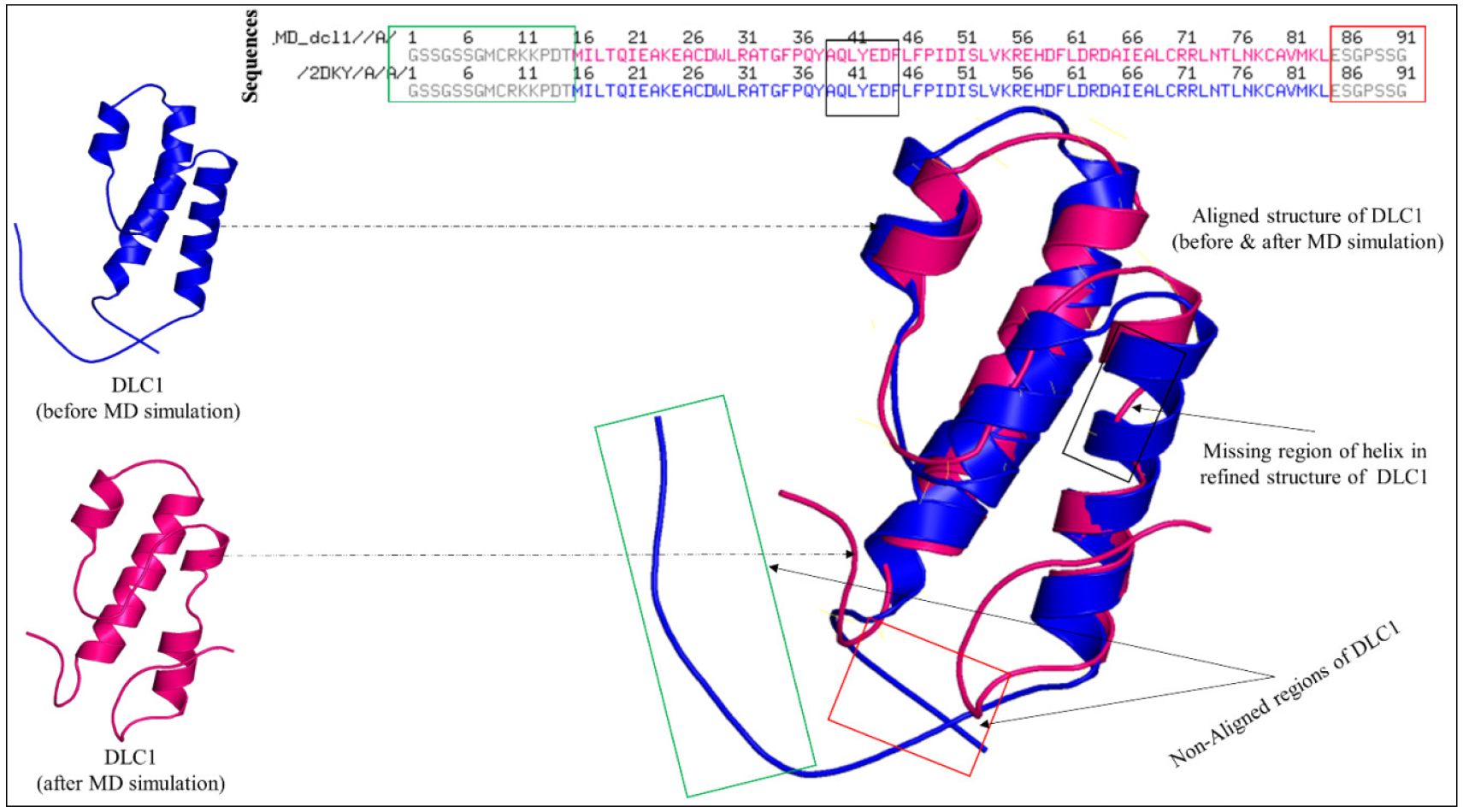
3.4. Stability Study of DLC1
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Necula, L.; Matei, L.; Dragu, D.; Neagu, A.; Mambet, C.; Nedeianu, S.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Recent advances in gastric cancer early diagnosis. World J. Gastroenterol. 2019, 25, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Globocan 2020. International Agency for Research on Cancer. Available online: https://gco.iarc.fr/today/data/factsheets/populations/900-world-fact-sheets.pdf (accessed on 10 January 2022).
- Hu, B.; El Hajj, N.; Sittler, S.; Lammert, N.; Barnes, R.; Meloni-Ehrig, A. Gastric cancer: Classification, histology and application of molecular pathology. J. Gastrointest. Oncol. 2012, 3, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Berlth, F.; Bollschweiler, E.; Drebber, U.; Hoelscher, A.H.; Moenig, S. Pathohistological classification systems in gastric cancer: Diagnostic relevance and prognostic value. World J. Gastroenterol. 2014, 20, 5679–5684. [Google Scholar] [CrossRef] [PubMed]
- Sitarz, R.; Skierucha, M.; Mielko, J.; Offerhaus, G.J.A.; Maciejewski, R.; Polkowski, W.P. Gastric cancer: Epidemiology, prevention, classification, and treatment. Cancer Manag. Res. 2018, 10, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, R.; Sharma, P.C. Next generation sequencing-based emerging trends in molecular biology of gastric cancer. Am. J. Cancer Res. 2018, 8, 207–225. [Google Scholar] [PubMed]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat Rev Cancer. 2010, 10, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Nagini, S. Carcinoma of the stomach: A review of epidemiology, pathogenesis, molecular genetics and chemoprevention. World J. Gastrointest. Oncol. 2012, 4, 156–169. [Google Scholar] [CrossRef]
- Pasechnikov, V.; Chukov, S.; Fedorov, E.; Kikuste, I.; Leja, M. Gastric cancer: Prevention, screening and early diagnosis. World J. Gastroenterol. 2014, 20, 13842–13862. [Google Scholar] [CrossRef]
- Hallinan, J.T.; Venkatesh, S.K. Gastric carcinoma: Imaging diagnosis, staging and assessment of treatment response. Cancer Imaging 2013, 13, 212–227. [Google Scholar] [CrossRef] [Green Version]
- Maconi, G.; Manes, G.; Porro, G.B. Role of symptoms in diagnosis and outcome of gastric cancer. World J. Gastroenterol. 2008, 14, 1149–1155. [Google Scholar] [CrossRef]
- Chong, X.; Peng, R.; Sun, Y.; Zhang, L.; Zhang, Z. Identification of Key Genes in Gastric Cancer by Bioinformatics Analysis. Biomed. Res. Int. 2020, 2020, 7658230. [Google Scholar] [CrossRef]
- Jabeen, A.; Ahmad, N.; Raza, K. Differential expression analysis of ZIKV infected human RNA sequence reveals potential genetic biomarkers. In Proceedings of the International Work-Conference on Bioinformatics and Biomedical Engineering, Granada, Spain, 8 May 2019; Springer: Cham, Switzerland, 2019; pp. 283–294. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Sirén, J.; Välimäki, N.; Mäkinen, V. Indexing Graphs for Path Queries with Applications in Genome Research. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 375–388. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Raza, K. Reconstruction, topological and gene ontology enrichment analysis of cancerous gene regulatory network modules. Curr. Bioinform. 2016, 11, 243–258. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Path towards comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, J.V.; Bernardi, R.C.; Rudack, T.; Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Schulten, K. QwikMD — Integrative Molecular Dynamics Toolkit for Novices and Experts. Sci. Rep. 2016, 6, 26536. [Google Scholar] [CrossRef] [Green Version]
- Anandakrishnan, R.; Drozdetski, A.; Walker, R.C.; Onufriev, A.V. Speed of Conformational Change: Comparing Explicit and Implicit Solvent Molecular Dynamics Simulations. Biophys. J. 2015, 108, 1153–1164. [Google Scholar] [CrossRef] [Green Version]
- Jenik, M.; Parra, R.G.; Radusky, L.G.; Turjanski, A.; Wolynes, P.G.; Ferreiro, D.U. Protein frustratometer: A tool to localize energetic frustration in protein molecules. Nucleic Acids Res. 2012, 40, W348–W351. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Lin, L.; Zhang, J.; Jiang, Y.; Pan, C.; Sun, L.; Duan, J.; Liao, W. Low expression of DLC1 is predictive of poor therapeutic efficiency of fluoropyrimidine and oxaliplatin as adjuvant chemotherapy in gastric cancer. Mol. Med. Rep. 2015, 12, 5771–5779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Cho, S.Y.; Kim, H.; Na, D.; Han, J.Y.; Chae, J.; Park, C.; Park, O.-K.; Min, S.; Kang, J.; et al. Genomic alterations in BCL2L1 and DLC1 contribute to drug sensitivity in gastric cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 12492–12497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Vascon, F.; Gasparotto, M.; Giacomello, M.; Cendron, L.; Bergantino, E.; Filippini, F.; Righetto, I. Protein electrostatics: From computational and structural analysis to discovery of functional fingerprints and biotechnological design. Comput. Struct. Biotechnol. J. 2020, 18, 1774–1789. [Google Scholar] [CrossRef] [PubMed]
- Qazi, S.; Raza, K. Phytochemicals from Ayurvedic plants as potential medicaments for ovarian cancer: An in silico analysis. J. Mol. Model. 2021, 27, 114. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Kim, T.Y.; Jong, H.S.; Song, S.H.; Dimtchev, A.; Jeong, S.-J.; Lee, J.W.; Kim, T.-Y.; Kim, N.K.; Jung, M.; Bang, Y.-J. Transcriptional silencing of the DLC-1 tumor suppressor gene by epigenetic mechanism in gastric cancer cells. Oncogene 2003, 22, 3943–3951. [Google Scholar] [CrossRef] [Green Version]
- Isabel, H. The Role of DLC1 in Helicobacter-Related Gastric Disease. Ph.D. Thesis, Ruperto-Carola University of Heidelberg, Heidelberg, Germany, 2019. Available online: https://archiv.ub.uni-heidelberg.de/volltextserver/25333/ (accessed on 10 June 2022).
- Liu, X.; Ma, B.; Chen, M.; Zhang, Y.; Ma, Z.; Chen, H. Prognostic Autophagy-Related Genes of Gastric Cancer Patients on Chemotherapy. Front. Genet. 2021, 12, 720849. [Google Scholar] [CrossRef]
- Facciorusso, A.; Del Prete, V.; Crucinio, N.; Serviddio, G.; Vendemiale, G.; Muscatiello, N. Lymphocyte-to-monocyte ratio predicts survival after radiofrequency ablation for colorectal liver metastases. World J. Gastroenterol. 2016, 22, 4211–4218. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment Accession No. | Sample Type | Total Bases (in Millions) |
|---|---|---|
| SRX11368669 | Gastric cancer | 945.80 |
| SRX11368670 | Gastric cancer | 1457.514 |
| SRX11639389 | Gastric cancer | 8610.47 |
| SRX11639390 | Gastric cancer | 9061.85 |
| SRX11799736 | Gastric cancer | 898.46 |
| SRX12181742 | Gastric cancer | 820.53 |
| SRX12181743 | Gastric cancer | 1004.16 |
| SRX12181745 | Gastric cancer | 1325.47 |
| SRX6479200 | Gastric cancer | 2080.26 |
| SRX6479201 | Gastric cancer | 2386.86 |
| SRX3808892 | Normal control | 10,655.83 |
| SRX3808894 | Normal control | 13,557.01 |
| SRX3808896 | Normal control | 13,036.12 |
| SRX3808898 | Normal control | 14,177.99 |
| SRX3808900 | Normal control | 14,125.66 |
| SRX3808902 | Normal control | 13,982.29 |
| SRX9904379 | Normal control | 4659.08 |
| SRX9904380 | Normal control | 3183.77 |
| SRX9904381 | Normal control | 2834.01 |
| SRX9904382 | Normal control | 4591.04 |
| Category | Term | Gene Count | % | p-Value | Benjamini |
|---|---|---|---|---|---|
| GOTERM_BP_DIRECT | Cation Transmembrane Transport | 4 | 2.9 | 0.0022 | 1.0 |
| GOTERM_BP_DIRECT | DNA Replication | 5 | 3.6 | 0.0097 | 1.0 |
| GOTERM_BP_DIRECT | Regulation of cardiac rate by cardiac conduction | 3 | 2.2 | 0.0150 | 1.0 |
| GOTERM_BP_DIRECT | Membrane depolarization during atrial cardiac muscle cell action potential | 2 | 1.4 | 0.0160 | 1.0 |
| GOTERM_BP_DIRECT | Regulation of endothelial tube morphogenesis | 2 | 1.4 | 0.0210 | 1.0 |
| GOTERM_BP_DIRECT | Positive regulation of cell adhesion | 3 | 2.2 | 0.0220 | 1.0 |
| GOTERM_BP_DIRECT | Membrane depolarization during AV node cell action potential | 2 | 1.4 | 0.0270 | 1.0 |
| GOTERM_BP_DIRECT | Chromatin-mediated maintenance of transcription | 2 | 1.4 | 0.0470 | 1.0 |
| GOTERM_BP_DIRECT | Negative regulation of excitatory post-synaptic potential | 2 | 1.4 | 0.0470 | 1.0 |
| Category | Term | Gene Count | % | p-Value | Benjamini |
|---|---|---|---|---|---|
| UP_SEQ_FEATURE | Compositionally biased region: Ser-rich | 9 | 6.5 | 0.0024 | 1 |
| UP_SEQ_FEATURE | Compositionally biased region: Poly-Ser | 9 | 6.5 | 0.0056 | 1 |
| UP_SEQ_FEATURE | Region of interest: HA-stretch | 2 | 1.4 | 0.0110 | 1 |
| UP_SEQ_FEATURE | Splice variant | 54 | 39.1 | 0.0110 | 1 |
| UP_SEQ_FEATURE | Domain: Leucine-zipper | 4 | 2.9 | 0.0220 | 1 |
| UP_SEQ_FEATURE | Nucleotide phosphate-binding region: ATP | 11 | 8 | 0.0370 | 1 |
| UP_SEQ_FEATURE | Sequence variant | 76 | 55.1 | 0.0380 | 1 |
| UP_SEQ_FEATURE | Compositionally biased region: Gln-rich | 4 | 2.9 | 0.0470 | 1 |
| Category | Term | Gene Count | % | p-Value | Benjamini |
|---|---|---|---|---|---|
| UP_KEYWORDS | Alternative splicing | 71 | 51.4 | 0.0024 | 0.4800 |
| UP_KEYWORDS | Transferase | 18 | 13.0 | 0.0069 | 0.5100 |
| UP_KEYWORDS | Coiled-coil | 26 | 18.8 | 0.0120 | 0.5100 |
| UP_KEYWORDS | ATP-binding | 15 | 10.9 | 0.0130 | 0.5100 |
| UP_KEYWORDS | Polymorphism | 75 | 54.3 | 0.0150 | 0.5100 |
| UP_KEYWORDS | Cytoskeleton | 13 | 9.4 | 0.0150 | 0.5100 |
| UP_KEYWORDS | Nucleotide-binding | 17 | 12.3 | 0.0220 | 0.6300 |
| UP_KEYWORDS | Cell junction | 9 | 6.5 | 0.0250 | 0.6300 |
| UP_KEYWORDS | Phosphoprotein | 54 | 39.1 | 0.0290 | 0.6600 |
| UP_KEYWORDS | Obesity | 3 | 2.2 | 0.0330 | 0.6800 |
| UP_KEYWORDS | Brugada syndrome | 2 | 1.4 | 0.0500 | 0.9400 |
| DLC1 | Accuracy | Generalized Born Self-Energy (kJ) | Coulomb Energy (kJ) | Electrostatic Solvation Energy (kJ/mol) | Solvent Accessible Surface Area (SASA) (A2) | Total Energy (kJ/mol) | RMSD | ROG |
| 0.88 | −10,212.9680 | −24,329.9612 | −6586.7651 | 5535.538 | −28,373.0488 | 0.59 ± 0.34 | 12.92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Bhat, A.M.; Qazi, S.; Raza, K. DLC1 as Druggable Target for Specific Subsets of Gastric Cancer: An RNA-seq-Based Study. Medicina 2023, 59, 514. https://doi.org/10.3390/medicina59030514
Yang L, Bhat AM, Qazi S, Raza K. DLC1 as Druggable Target for Specific Subsets of Gastric Cancer: An RNA-seq-Based Study. Medicina. 2023; 59(3):514. https://doi.org/10.3390/medicina59030514
Chicago/Turabian StyleYang, Lianlei, Adil Manzoor Bhat, Sahar Qazi, and Khalid Raza. 2023. "DLC1 as Druggable Target for Specific Subsets of Gastric Cancer: An RNA-seq-Based Study" Medicina 59, no. 3: 514. https://doi.org/10.3390/medicina59030514