
Predictive Value of Gene Databases in Discovering New Biomarkers and New Therapeutic Targets in Lung Cancer
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection and Processing
2.2. Identification of Differential Genes
2.3. Prediction of ICI Using Single-Sample Gene Set Enrichment Analysis (ssGSEA)
2.4. Establishment of Weighted Gene Co-Expression Network Analysis (WGCNA)
2.5. Identification of Immune Cell-Associated Differential Genes
2.6. Functional Enrichment and Pathway Analysis of Immune Cell-Associated DEGs
2.7. Protein-Protein Interaction (PPI) Network and Hub Gene Identification
2.8. Hub Gene Expression and Its Diagnostic Performance
2.9. Correlation and Survival Analysis of Hub Genes with Tumor-Infiltrating Immune Cells
3. Results
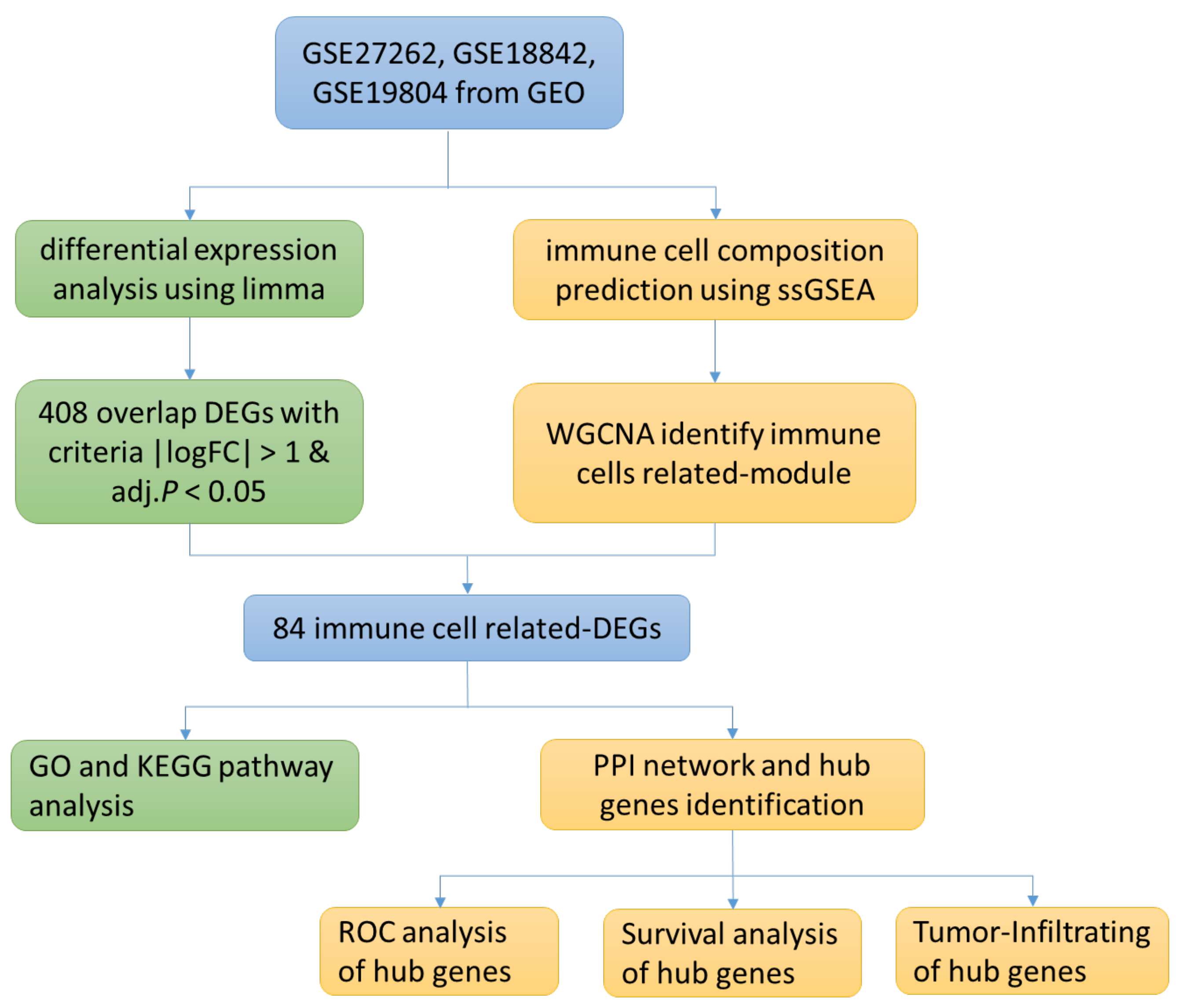
3.1. Research Flow Chart
3.2. Differential Gene Identification
3.3. Identification of Immune Cell-Related Module Genes by ssGSEA and WGCNA
3.4. Functional Enrichment Analysis of Immune Cell-Associated Differential Genes
3.5. Identification of Hub Genes and ROC Curve Analysis
3.6. Correlation and Survival Analysis of Hub Gene and Tumor-Infiltrating Immune Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Romaszko, A.M.; Doboszyńska, A. Multiple primary lung cancer: A literature review. Adv. Clin. Exp. Med. 2018, 27, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, L.; Zhou, C. Lung cancer in China: Current and prospect. Curr. Opin. Oncol. 2021, 33, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Bade, B.C.; Cruz, C.D. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest. Med. 2020, 41, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, Z.; Zhao, Y.; Zhang, Q.; Wu, X.; Miao, B.; Cao, J.; Fei, S. FN1, SPARC, and SERPINE1 are highly expressed and significantly related to a poor prognosis of gastric adenocarcinoma revealed by microarray and bioinformatics. Sci. Rep. 2019, 9, 7827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Jin, Y.; Chen, Y.; Yao, H.B.; Xia, Y.J.; Ma, Y.Y.; Wang, W.; Shao, Q.S. Downregulation of ALDOB is associated with poor prognosis of patients with gastric cancer. Onco Targets Ther. 2016, 9, 6099–6109. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Ma, X.; Ha, W. Identification of Potential Prognostic Biomarkers Associated With Macrophage M2 Infiltration in Gastric Cancer. Front. Genet. 2021, 12, 827444. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yuan, Q.; Wu, D.; Meng, X.; Wang, B. Identification of core genes and outcome in gastric cancer using bioinformatics analysis. Oncotarget 2017, 8, 70271–70280. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Fu, T.; Dai, L.J.; Wu, S.Y.; Xiao, Y.; Ma, D.; Jiang, Y.Z.; Shao, Z.M. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J. Hematol. Oncol. 2021, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Pio, R.; Ajona, D.; Ortiz-Espinosa, S.; Mantovani, A.; Lambris, J.D. Complementing the Cancer-Immunity Cycle. Front. Immunol. 2019, 10, 774. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Pinello, N.; Sun, S.; Wong, J. Aberrant expression of enzymes regulating m(6)A mRNA methylation: Implication in cancer. Cancer Biol. Med. 2018, 15, 323–334. [Google Scholar] [PubMed] [Green Version]
- Tong, J.; Cao, G.; Zhang, T.; Sefik, E.; Amezcua Vesely, M.C.; Broughton, J.P.; Zhu, S.; Li, H.; Li, B.; Chen, L.; et al. m(6)A mRNA methylation sustains Treg suppressive functions. Cell Res. 2018, 28, 253–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Yu, M.; Xu, X.; Wang, Y.; Xing, H.; An, J.; Yang, J.; Tang, C.; Sun, D.; Zhu, Y. Identification of biomarkers related to CD8(+) T cell infiltration with gene co-expression network in clear cell renal cell carcinoma. Aging 2020, 12, 3694–3712. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhang, G. Identification of immune-infiltrating cell-related biomarkers in hepatocellular carcinoma based on gene co-expression network analysis. Diagn. Pathol. 2021, 16, 57. [Google Scholar] [CrossRef]
- Tian, Z.; He, W.; Tang, J.; Liao, X.; Yang, Q.; Wu, Y.; Wu, G. Identification of Important Modules and Biomarkers in Breast Cancer Based on WGCNA. Onco Targets Ther. 2020, 13, 6805–6817. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, N.J.; Bensen, J.T.; Poole, C.; Troester, M.A.; Gammon, M.D.; Luo, J.; Millikan, R.C.; Olshan, A.F. Genetic variation in cell cycle regulatory gene AURKA and association with intrinsic breast cancer subtype. Mol. Carcinog. 2015, 54, 1668–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, S.L.; Fenaux, P.; Craig, M.D.; Gyan, E.; Lister, J.; Kassis, J.; Pigneux, A.; Schiller, G.J.; Jung, J.; Leonard, E.J.; et al. An exploratory phase 2 study of investigational Aurora A kinase inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leuk. Res. Rep. 2014, 3, 58–61. [Google Scholar] [CrossRef] [Green Version]
- Wang-Bishop, L.; Chen, Z.; Gomaa, A.; Lockhart, A.C.; Salaria, S.; Wang, J.; Lewis, K.B.; Ecsedy, J.; Washington, K.; Beauchamp, R.D.; et al. Inhibition of AURKA Reduces Proliferation and Survival of Gastrointestinal Cancer Cells With Activated KRAS by Preventing Activation of RPS6KB1. Gastroenterology 2019, 156, 662–675.e7. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lyu, J.; Yang, E.J.; Liu, Y.; Zhang, B.; Shim, J.S. Targeting AURKA-CDC25C axis to induce synthetic lethality in ARID1A-deficient colorectal cancer cells. Nat. Commun. 2018, 9, 3212. [Google Scholar] [CrossRef]
- Donnella, H.J.; Webber, J.T.; Levin, R.S.; Camarda, R.; Momcilovic, O.; Bayani, N.; Shah, K.N.; Korkola, J.E.; Shokat, K.M.; Goga, A.; et al. Kinome rewiring reveals AURKA limits PI3K-pathway inhibitor efficacy in breast cancer. Nat. Chem. Biol. 2018, 14, 768–777. [Google Scholar] [CrossRef]
- Guo, M.; Lu, S.; Huang, H.; Wang, Y.; Yang, M.Q.; Yang, Y.; Fan, Z.; Jiang, B.; Deng, Y. Increased AURKA promotes cell proliferation and predicts poor prognosis in bladder cancer. BMC Syst. Biol. 2018, 12 (Suppl. 7), 118. [Google Scholar] [CrossRef]
- Schneider, M.A.; Christopoulos, P.; Muley, T.; Warth, A.; Klingmueller, U.; Thomas, M.; Herth, F.J.; Dienemann, H.; Mueller, N.S.; Theis, F.; et al. AURKA, DLGAP5, TPX2, KIF11 and CKAP5: Five specific mitosis-associated genes correlate with poor prognosis for non-small cell lung cancer patients. Int. J. Oncol. 2017, 50, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.Y.; Liu, X.X.; Li, H.; Li, R.; Liu, X.; Qu, Y.Q. Elevated mRNA Levels of AURKA, CDC20 and TPX2 are associated with poor prognosis of smoking related lung adenocarcinoma using bioinformatics analysis. Int. J. Med. Sci. 2018, 15, 1676–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, K.N.; Bhatt, R.; Rotow, J.; Rohrberg, J.; Olivas, V.; Wang, V.E.; Hemmati, G.; Martins, M.M.; Maynard, A.; Kuhn, J.; et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Al-Khafaji, A.S.K.; Marcus, M.W.; Davies, M.P.A.; Risk, J.M.; Shaw, R.J.; Field, J.K.; Liloglou, T. AURKA mRNA expression is an independent predictor of poor prognosis in patients with non-small cell lung cancer. Oncol. Lett. 2017, 13, 4463–4468. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, E.O.; Carneiro-Lobo, T.C.; Aoki, M.N.; Levantini, E.; Bassères, D.S. Aurora kinase targeting in lung cancer reduces KRAS-induced transformation. Mol. Cancer 2016, 15, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Huo, C.; Jiang, Y.; Liu, J.; Yang, Y.; Yin, Y.; Qu, Y. AURKA and FAM83A are prognostic biomarkers and correlated with Tumor-infiltrating Lymphocytes in smoking related Lung Adenocarcinoma. J. Cancer 2021, 12, 1742–1754. [Google Scholar] [CrossRef]
- Wang, J.; Hu, T.; Wang, Q.; Chen, R.; Xie, Y.; Chang, H.; Cheng, J. Repression of the AURKA-CXCL5 axis induces autophagic cell death and promotes radiosensitivity in non-small-cell lung cancer. Cancer Lett. 2021, 509, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Li, W.; Zou, Z.; Zou, X.; Wang, C. CCNB1 is a prognostic biomarker for ER+ breast cancer. Med. Hypotheses 2014, 83, 359–364. [Google Scholar] [CrossRef]
- El-Huneidi, W.; Shehab, N.G.; Bajbouj, K.; Vinod, A.; El-Serafi, A.; Shafarin, J.; Bou Malhab, L.J.; Abdel-Rahman, W.M.; Abu-Gharbieh, E. Micromeria fruticosa Induces Cell Cycle Arrest and Apoptosis in Breast and Colorectal Cancer Cells. Pharmaceuticals 2020, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Bomont, P.; Maddox, P.; Shah, J.V.; Desai, A.B.; Cleveland, D.W. Unstable microtubule capture at kinetochores depleted of the centromere-associated protein CENP-F. Embo J. 2005, 24, 3927–3939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Liu, X.; Liu, T.; Meng, X.; Yin, X.; Fang, C.; Huang, D.; Cao, Y.; Weng, H.; Zeng, X.; et al. Identification of Biomarkers Correlated with the TNM Staging and Overall Survival of Patients with Bladder Cancer. Front. Physiol. 2017, 8, 947. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Huang, J.; Lan, J.; Zhou, K.; Gao, Y.; Song, Z.; Deng, Y.; Liu, L.; Dong, Y.; Liu, X. Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell Int. 2019, 19, 264. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.L.; Fagan, A.; Fox, E.J.; Millikan, R.C.; Culhane, A.C.; Brennan, D.J.; McCann, A.H.; Hegarty, S.; Moyna, S.; Duffy, M.J.; et al. CENP-F expression is associated with poor prognosis and chromosomal instability in patients with primary breast cancer. Int. J. Cancer 2007, 120, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Liu, L.; Zeng, T.; Zhu, Y.H.; Li, J.; Chen, L.; Li, Y.; Yuan, Y.F.; Ma, S.; Guan, X.Y. Characterization of the oncogenic function of centromere protein F in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2013, 436, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.J.; Gao, J.; Xie, J.D.; Cao, J.Y.; Cui, S.X.; Gao, H.J.; Yao, S.P.; Liu, T.; Zhang, Y.Y.; Guo, C.H.; et al. Prognostic relevance and therapeutic implications of centromere protein F expression in patients with esophageal squamous cell carcinoma. Dis. Esophagus 2013, 26, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Liu, L.; Chen, S.P.; Zhang, X.; Mi, Y.J.; Liu, Z.G.; Li, M.Z.; Zhang, H.; Qian, C.N.; Shao, J.Y.; et al. Prognostic significance and therapeutic implications of centromere protein F expression in human nasopharyngeal carcinoma. Mol. Cancer 2010, 9, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andriani, F.; Roz, E.; Caserini, R.; Conte, D.; Pastorino, U.; Sozzi, G.; Roz, L. Inactivation of both FHIT and p53 cooperate in deregulating proliferation-related pathways in lung cancer. J. Thorac. Oncol. 2012, 7, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.P.; Luo, R. MLF1IP is correlated with progression and prognosis in luminal breast cancer. Biochem. Biophys. Res. Commun. 2016, 477, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Therkildsen, C.; Veerla, S.; Jönsson, M.; Bernstein, I.; Borg, A.; Nilbert, M. Distinct gene expression signatures in lynch syndrome and familial colorectal cancer type x. PLoS ONE 2013, 8, e71755. [Google Scholar] [CrossRef]
- Wang, S.; Liu, B.; Zhang, J.; Sun, W.; Dai, C.; Sun, W.; Li, Q. Centromere protein U is a potential target for gene therapy of human bladder cancer. Oncol. Rep. 2017, 38, 735–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons-Evelyn, M.; Bailey-Dell, K.; Toretsky, J.A.; Ross, D.D.; Fenton, R.; Kalvakolanu, D.; Rapoport, A.P. PBK/TOPK is a novel mitotic kinase which is upregulated in Burkitt’s lymphoma and other highly proliferative malignant cells. Blood Cells Mol. Dis. 2001, 27, 825–829. [Google Scholar] [CrossRef]
- Zhao, S.; Dai, J.; Zhao, W.; Xia, F.; Zhou, Z.; Wang, W.; Gu, S.; Ying, K.; Xie, Y.; Mao, Y. PDZ-binding kinase participates in spermatogenesis. Int. J. Biochem. Cell Biol. 2001, 33, 631–636. [Google Scholar] [CrossRef]
- Ma, H.; Li, Y.; Wang, X.; Wu, H.; Qi, G.; Li, R.; Yang, N.; Gao, M.; Yan, S.; Yuan, C.; et al. PBK, targeted by EVI1, promotes metastasis and confers cisplatin resistance through inducing autophagy in high-grade serous ovarian carcinoma. Cell Death Dis. 2019, 10, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Xu, S. PBK/TOPK overexpression and survival in solid tumors: A PRISMA-compliant meta-analysis. Medicine 2019, 98, e14766. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Liu, Z.G.; Du, K.Y.; Wu, J.H.; Yang, N.; Malhotra, A.; Shu, J.K. Potential of β-elemene induced ferroptosis through Pole2-mediated p53 and PI3K/AKT signaling in lung cancer cells. Chem. Biol. Interact. 2022, 365, 110088. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, P.; Ma, X.; Zeng, S.; Peng, Y.; Zhang, G. Therapeutic restoring p53 function with small molecule for oncogene-driven non-small cell lung cancer by targeting serine 392 phosphorylation. Biochem. Pharmacol. 2022, 203, 115188. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO | Platform | Tumor | Normal | DEGs |
|---|---|---|---|---|
| GSE27262 | GPL570 | 25 | 25 | 562 |
| GSE18842 | GPL570 | 46 | 45 | 2568 |
| GSE19804 | GPL570 | 60 | 60 | 1197 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Yu, X.; Qu, C.; Xu, S. Predictive Value of Gene Databases in Discovering New Biomarkers and New Therapeutic Targets in Lung Cancer. Medicina 2023, 59, 547. https://doi.org/10.3390/medicina59030547
Liu M, Yu X, Qu C, Xu S. Predictive Value of Gene Databases in Discovering New Biomarkers and New Therapeutic Targets in Lung Cancer. Medicina. 2023; 59(3):547. https://doi.org/10.3390/medicina59030547
Chicago/Turabian StyleLiu, Mengfeng, Xiran Yu, Changfa Qu, and Shidong Xu. 2023. "Predictive Value of Gene Databases in Discovering New Biomarkers and New Therapeutic Targets in Lung Cancer" Medicina 59, no. 3: 547. https://doi.org/10.3390/medicina59030547