
Alzheimer’s Disease Treatment: The Search for a Breakthrough

 ,
, 
 , ,
, , 
Abstract
:1. Introduction
2. Finding a Viable Approach
3. Inflammation in AD
3.1. Overview
3.2. Neuroinflammation and Microglia
3.3. Anti-Inflammatory Drug Repurposing as an Approach to AD via Microglia
3.4. Repurposing Anti-TNF Agents
3.5. Inciting the M2/TREM 2 Phenotype in Microglia
3.6. CD33
3.7. PTI-125
3.8. Role of Peripheral Inflammation in AD
4. Delivery Systems to the Brain Crossing the BBB
5. Stem Cells
6. Deep Brain Stimulation
7. Diet as a Preventative Measure
7.1. Overall Dietary Pattern
7.2. Calorie Restriction
7.3. Vitamin D
7.4. The B Vitamins: B6 (Pyridoxine), Folate (B9), B12 (Cobalamin)
7.5. Antioxidants
8. Mental and Physical Activity
8.1. Exercise and Physical Activity
8.2. Mental Exercise
9. The Future
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Rabinovici, G.D. Late-onset Alzheimer Disease. Continuum 2019, 25, 14–33. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fändrich, M.; et al. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. Contributors. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Wimo, A.; Guerchet, M.; Ali, G.C.; Wu, Y.T.; Prina, A.M.; Winblad, B.; Jönsson, L.; Liu, Z.; Prince, M. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimer’s Dement. 2017, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Volloch, V.; Rits-Volloch, S. Effect of Lecanemab in Early Alzheimer’s Disease: Mechanistic Interpretation in the Amyloid Cascade Hypothesis 2.0 Perspective. J. Alzheimers Dis. 2023. [Google Scholar] [CrossRef]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Lecanemab: First Approval. Drugs 2023, 83, 359–365. [Google Scholar] [CrossRef]
- Daly, T.; Epelbaum, S. The Accelerated Approval of Aducanumab Invites a Rethink of the Current Model of Drug Development for Alzheimer’s Disease. AJOB Neurosci. 2022, 1–4. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M. Drug trial for Alzheimer’s disease is a game changer. Nature 2023, 615, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.; Jaqua, E.; Safaeipour, M.; Ladue, T. Conventional Versus New Treatment: Comparing the Effects of Acetylcholinesterase Inhibitors and N-Methyl-D-Aspartate Receptor Antagonist with Aducanumab. Cureus 2022, 14, e31065. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Suh, C.H.; Shim, W.H.; Lim, J.S.; Lee, J.H.; Kim, S.J. Incidence of Amyloid-Related Imaging Abnormalities in Patients with Alzheimer Disease Treated With Anti-β-Amyloid Immunotherapy: A Meta-analysis. Neurology 2022, 99, e2092–e2101. [Google Scholar] [CrossRef] [PubMed]
- Villain, N.; Planche, V.; Levy, R. High-clearance anti-amyloid immunotherapies in Alzheimer’s disease. Part 1: Meta-analysis and review of efficacy and safety data, and medico-economical aspects. Rev. Neurol. 2022, 178, 1011–1030. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Cummings, J.; Schobel, S.; Salloway, S.; Vellas, B.; Boada, M.; Black, S.E.; Blennow, K.; Fontoura, P.; Klein, G.; et al. Gantenerumab: An anti-amyloid monoclonal antibody with potential disease-modifying effects in early Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 178. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Lessons from antiamyloid-beta immunotherapies in Alzheimer’s disease. Handb. Clin. Neurol. 2023, 193, 267–292. [Google Scholar] [CrossRef]
- Reiss, A.B.; Montufar, N.; DeLeon, J.; Pinkhasov, A.; Gomolin, I.H.; Glass, A.D.; Arain, H.A.; Stecker, M.M. Alzheimer Disease Clinical Trials Targeting Amyloid: Lessons Learned from Success in Mice and Failure in Humans. Neurologist 2021, 26, 52–61. [Google Scholar] [CrossRef]
- Reiss, A.B.; Glass, A.D.; Wisniewski, T.; Wolozin, B.; Gomolin, I.H.; Pinkhasov, A.; De Leon, J.; Stecker, M.M. Alzheimer’s disease: Many failed trials, so where do we go from here? J. Investig. Med. 2020, 68, 1135–1140. [Google Scholar] [CrossRef]
- Kim, C.K.; Lee, Y.R.; Ong, L.; Gold, M.; Kalali, A.; Sarkar, J. Alzheimer’s Disease: Key Insights from Two Decades of Clinical Trial Failures. J. Alzheimer’s Dis. 2022, 87, 83–100. [Google Scholar] [CrossRef]
- Jackson, J.; Jambrina, E.; Li, J.; Marston, H.; Menzies, F.; Phillips, K.; Gilmour, G. Targeting the Synapse in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 735. [Google Scholar] [CrossRef] [Green Version]
- De Wilde, M.C.; Overk, C.R.; Sijben, J.W.; Masliah, E. Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimer’s Dement. 2016, 12, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Henstridge, C.M.; Hyman, B.T.; Spires-Jones, T.L. Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 2019, 20, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Nunomura, A.; Hirai, K.; Zhu, X.; Pérez, M.; Avila, J.; Castellani, R.J.; Atwood, C.S.; Aliev, G.; Sayre, L.M.; et al. Is Oxidative Damage the Fundamental Pathogenic Mechanism of Alzheimer’s and Other Neurodegenerative Diseases? Free Radic. Biol. Med. 2002, 33, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Cummings, J.; Jack, C.R., Jr.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimers Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D. Controversy and Progress in Alzheimer’s Disease—FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Howard, R.; McShane, R.; Lindesay, J.; Ritchie, C.; Baldwin, A.; Barber, R.; Burns, A.; Dening, T.; Findlay, D.; Holmes, C.; et al. Donepezil and Memantine for Moderate-to-Severe Alzheimer’s disease. N. Engl. J. Med. 2012, 366, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Johnson, H.J.; Koshy, A.A. Understanding neuroinflammation through central nervous system infections. Curr. Opin. Neurobiol. 2022, 76, 102619. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meraz-Ríos, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernández, J.; Campos-Peña, V. Inflammatory process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combs, C.K.; Johnson, D.E.; Cannady, S.B.; Lehman, T.M.; Landreth, G.E. Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J. Neurosci. 1999, 19, 928–939. [Google Scholar] [CrossRef] [Green Version]
- Sarma, J.D. Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J. Neurovirol. 2014, 20, 122–136. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C. The influence of environment and origin on brain resident macrophages and implications for therapy. Nat. Neurosci. 2019, 23, 157–166. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo, P.; Attwell, D.; Madry, C. Ion Channels and Receptors as Determinants of Microglial Function. Trends Neurosci. 2019, 42, 278–292. [Google Scholar] [CrossRef]
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.E. Dynamics of the Microglial/Amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 2008, 28, 4283–4292. [Google Scholar] [CrossRef] [Green Version]
- Wyss-Coray, T.; Yan, F.; Lin, A.H.-T.; Lambris, J.D.; Alexander, J.J.; Quigg, R.J. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc. Natl. Acad. Sci. USA 2002, 99, 10837–10842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Son, S.M.; Mook-Jung, I. Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 2016, 64, 2274–2290. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—New prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Zhang, B.; Wei, Y.Z.; Wang, G.Q.; Li, D.D.; Shi, J.S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell Neurosci. 2018, 12, 531. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef] [PubMed]
- Jordan, F.; Quinn, T.J.; McGuinness, B.; Passmore, P.; Kelly, J.P.; Tudur Smith, C.; Murphy, K.; Devane, D. Aspirin and other non-steroidal anti-inflammatory drugs for the prevention of dementia. Cochrane Database Syst. Rev. 2020, 4, CD011459. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Ghosh, T. Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer’s Disease. J. Neuroimmunol. 2016, 291, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.B.; Rasmusson, D.X.; Folstein, M.F.; Carson, K.A.; Kawas, C.; Brandt, J. Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology 1995, 45, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Beard, C.M.; Waring, S.C.; O’Brien, P.C.; Kurland, L.T.; Kokmen, E. Nonsteroidal anti-inflammatory drug use and Alzheimer’s disease: A case-control study in Rochester, Minnesota, 1980 through 1984. Mayo Clin. Proc. 1998, 73, 951–955. [Google Scholar] [CrossRef]
- Miguel-Álvarez, M.; Santos-Lozano, A.; Sanchis-Gomar, F.; Fiuza-Luces, C.; Pareja-Galeano, H.; Garatachea, N.; Lucia, A. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: A systematic review and meta-analysis of treatment effect. Drugs Aging 2015, 32, 139–147. [Google Scholar] [CrossRef]
- Stewart, W.F.; Kawas, C.; Corrada, M.; Metter, E.J. Risk of Alzheimer’s disease and duration of NSAID use. Neurology 1997, 48, 626–632. [Google Scholar] [CrossRef]
- Meyer, P.F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.É.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2019, 92, e2070–e2080. [Google Scholar] [CrossRef] [Green Version]
- Desai, R.J.; Varma, V.R.; Gerhard, T.; Segal, J.; Mahesri, M.; Chin, K.; Horton, D.B.; Kim, S.C.; Schneeweiss, S.; Thambisetty, M. Comparative Risk of Alzheimer Disease and Related Dementia Among Medicare Beneficiaries with Rheumatoid Arthritis Treated With Targeted Disease-Modifying Antirheumatic Agents. JAMA Netw. Open 2022, 5, e226567. [Google Scholar] [CrossRef]
- Li, F.; Eteleeb, A.M.; Buchser, W.; Sohn, C.; Wang, G.; Xiong, C.; Payne, P.R.; McDade, E.; Karch, C.M.; Harari, O.; et al. Weakly activated core neuroinflammation pathways were identified as a central signaling mechanism contributing to the chronic neurodegeneration in Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 935279. [Google Scholar] [CrossRef]
- Torres-Acosta, N.; O’Keefe, J.H.; O’Keefe, E.L.; Isaacson, R.; Small, G. Therapeutic potential of TNF-alpha inhibition for Alzheimer’s disease prevention. J. Alzheimer’s Dis. 2020, 78, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Ortí-Casañ, N.; Wu, Y.; Naudé, P.J.W.; De Deyn, P.P.; Zuhorn, I.S.; Eisel, U.L.M. Targeting TNFR2 as a Novel Therapeutic Strategy for Alzheimer’s Disease. Front. Neurosci. 2019, 13, 49. [Google Scholar] [CrossRef] [Green Version]
- Detrait, E.R.; Danis, B.; Lamberty, Y.; Foerch, P. Peripheral administration of an anti-TNF-alpha receptor fusion protein counteracts the amyloid induced elevation of hippocampal TNF-alpha levels and memory deficits in mice. Neurochem. Int. 2014, 72, 10–13. [Google Scholar] [CrossRef]
- Li, Y.; Fan, H.; Ni, M.; Zhang, W.; Fang, F.; Sun, J.; Lyu, P.; Ma, P. Etanercept Reduces Neuron Injury and Neuroinflammation via Inactivating c-Jun N-terminal Kinase and Nuclear Factor-κB Pathways in Alzheimer’s Disease: An In Vitro and In Vivo Investigation. Neuroscience 2022, 484, 140–150. [Google Scholar] [CrossRef]
- Shi, J.Q.; Shen, W.; Chen, J.; Wang, B.R.; Zhong, L.L.; Zhu, Y.W.; Zhu, H.Q.; Zhang, Q.Q.; Zhang, Y.D.; Xu, J. Anti-TNF-α reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011, 1368, 239–247. [Google Scholar] [CrossRef]
- Barnum, C.J.; Chen, X.; Chung, J.; Chang, J.; Williams, M.; Grigoryan, N.; Tesi, R.J.; Tansey, M.G. Peripheral administration of the selective inhibitor of soluble tumor necrosis factor (TNF) XPro®1595 attenuates nigral cell loss and glial activation in 6-OHDA hemiparkinsonian rats. J. Park. Dis. 2014, 4, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacPherson, K.P.; Sompol, P.; Kannarkat, G.T.; Chang, J.; Sniffen, L.; Wildner, M.E.; Norris, C.M.; Tansey, M.G. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 2017, 102, 81–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbimbo, B.P.; Ippati, S.; Watling, M. Should drug discovery scientists still embrace the amyloid hypothesis for Alzheimer’s disease or should they be looking elsewhere? Expert Opin. Drug Discov. 2020, 15, 1241–1251. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Remolina Serrano, J.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef] [Green Version]
- Paloneva, J.; Manninen, T.; Christman, G.; Hovanes, K.; Mandelin, J.; Adolfsson, R.; Bianchin, M.; Bird, T.; Miranda, R.; Salmaggi, A.; et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 2002, 71, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, B.R.; Sudduth, T.L.; Weekman, E.M.; Johnson, S.; Hawthorne, D.; Woolums, A.; Wilcock, D.M. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J. Neuroinflamm. 2020, 17, 238. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef] [PubMed]
- Andronie-Cioara, F.L.; Ardelean, A.I.; Nistor-Cseppento, C.D.; Jurcau, A.; Jurcau, M.C.; Pascalau, N.; Marcu, F. Molecular Mechanisms of Neuroinflammation in Aging and Alzheimer’s Disease Progression. Int. J. Mol. Sci. 2023, 24, 1869. [Google Scholar] [CrossRef]
- Butler, C.A.; Thornton, P.; Brown, G.C. CD33M inhibits microglial phagocytosis, migration and proliferation, but the Alzheimer’s disease-protective variant CD33m stimulates phagocytosis and proliferation, and inhibits adhesion. J. Neurochem. 2021, 158, 297–310. [Google Scholar] [CrossRef]
- Raj, T.; Ryan, K.J.; Replogle, J.M.; Chibnik, L.B.; Rosenkrantz, L.; Tang, A.; Rothamel, K.; Stranger, B.E.; Bennett, D.A.; Evans, D.A.; et al. CD33: Increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum. Mol. Genet. 2014, 1523, 2729–2736. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.G.; Whetzel, A.M.; Serrano, G.; Sue, L.I.; Beach, T.G.; Lue, L.F. Association of CD33 polymorphism rs3865444 with Alzheimer’s disease pathology and CD33 expression in human cerebral cortex. Neurobiol. Aging 2015, 36, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Wißfeld, J.; Nozaki, I.; Mathews, M.; Raschka, T.; Ebeling, C.; Hornung, V.; Brüstle, O.; Neumann, H. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia 2021, 69, 1393–1412. [Google Scholar] [CrossRef]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Alzforum. Available online: https://www.alzforum.org/therapeutics/al003 (accessed on 20 March 2023).
- Miles, L.A.; Hermans, S.J.; Crespi, G.A.N.; Gooi, J.H.; Doughty, L.; Nero, T.L.; Markulić, J.; Ebneth, A.; Wroblowski, B.; Oehlrich, D.; et al. Small Molecule Binding to Alzheimer Risk Factor CD33 Promotes Aβ Phagocytosis. iScience 2019, 19, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Lee, K.C.; Pei, Z.; Khan, A.; Bakshi, K.; Burns, L.H. PTI-125 binds and reverses an altered conformation of filamin A to reduce Alzheimer’s disease pathogenesis. Neurobiol. Aging 2017, 55, 99–114. [Google Scholar] [CrossRef]
- Wang, H.Y.; Pei, Z.; Lee, K.C.; Lopez-Brignoni, E.; Nikolov, B.; Crowley, C.A.; Marsman, M.R.; Barbier, R.; Friedmann, N.; Burns, L.H. PTI-125 Reduces Biomarkers of Alzheimer’s Disease in Patients. J. Prev. Alzheimers Dis. 2020, 7, 256–264. [Google Scholar] [CrossRef]
- Piller, C. Research backing experimental Alzheimer’s drug was first target of suspicion. Science 2022, 377, 363. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, R.F.; Bos, M.J.; Portegies, M.L.; Hofman, A.; Franco, O.H.; Koudstaal, P.J.; Ikram, M.A. The potential for prevention of dementia across two decades: The prospective, population-based Rotterdam Study. BMC Med. 2015, 13, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukadam, N.; Sommerlad, A.; Huntley, J.; Livingston, G. Population attributable fractions for risk factors for dementia in low-income and middle-income countries: An analysis using cross-sectional survey data. Lancet Glob. Health 2019, 7, e596–e603. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Peters, S.A.; Woodward, M.; Mejia Arango, S.; Batty, G.D.; Beckett, N.; Beiser, A.; Borenstein, A.R.; Crane, P.K.; Haan, M.; et al. Type 2 Diabetes as a Risk Factor for Dementia in Women Compared with Men: A Pooled Analysis of 2.3 Million People Comprising More Than 100,000 Cases of Dementia. Diabetes Care 2016, 39, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Nianogo, R.A.; Rosenwohl-Mack, A.; Yaffe, K.; Carrasco, A.; Hoffmann, C.M.; Barnes, D.E. Risk factors associated with Alzheimer disease and related dementias by sex and race and ethnicity in the US. JAMA Neurol. 2022, 79, 584–591. [Google Scholar] [CrossRef]
- Femminella, G.D.; Livingston, N.R.; Raza, S.; van der Doef, T.; Frangou, E.; Love, S.; Busza, G.; Calsolaro, V.; Carver, S.; Holmes, C.; et al. Does insulin resistance influence neurodegeneration in non-diabetic Alzheimer’s subjects? Alzheimers Res. Ther. 2021, 13, 47. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Wands, J.R. Alzheimer’s Disease Is Type 3 Diabetes-Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Giau, V.V. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef] [PubMed]
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 Diabetes and its Impact on the Immune System. Curr. Diabetes Rev. 2020, 16, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.N.; Chorawala, M.R.; Shah, M.B.; Shah, K.C.; Dave, B.P.; Shah, M.P.; Patel, T.M. Emerging Pathophysiological Mechanisms Linking Diabetes Mellitus and Alzheimer’s Disease: An Old Wine in a New Bottle. J. Alzheimers Dis. Rep. 2022, 6, 349–357. [Google Scholar] [CrossRef]
- Chatterjee, S.; Mudher, A. Alzheimer’s Disease and Type 2 Diabetes: A Critical Assessment of the Shared Pathological Traits. Front. Neurosci. 2018, 12, 383. [Google Scholar] [CrossRef] [Green Version]
- Swaroop, J.J.; Rajarajeswari, D.; Naidu, J.N. Association of TNF-α with insulin resistance in type 2 diabetes mellitus. Indian J. Med. Res. 2012, 135, 127–130. [Google Scholar] [CrossRef]
- Elahi, M.; Hasan, Z.; Motoi, Y.; Matsumoto, S.E.; Ishiguro, K.; Hattori, N. Region-Specific Vulnerability to Oxidative Stress, Neuroinflammation, and Tau Hyperphosphorylation in Experimental Diabetes Mellitus Mice. J. Alzheimers Dis. 2016, 51, 1209–1224. [Google Scholar] [CrossRef]
- Piątkowska-Chmiel, I.; Herbet, M.; Gawrońska-Grzywacz, M.; Dudka, J. Regulation of Neuroinflammatory Signaling by PPARγ Agonist in Mouse Model of Diabetes. Int. J. Mol. Sci. 2022, 23, 5502. [Google Scholar] [CrossRef]
- Jung, H.J.; Park, S.S.; Mok, J.O.; Lee, T.K.; Park, C.S.; Park, S.A. Increased expression of three-repeat isoforms of tau contributes to tau pathology in a rat model of chronic type 2 diabetes. Exp. Neurol. 2011, 228, 232–241. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.Y.; Liu, S.F.; Guo, Y.G.; Ma, Z.Q.; Li, Y.; Wang, S.J.; Niu, Y.; Li, M.; Zhai, J.J.; Shang, S.H.; et al. Diabetes influences the fusion of autophagosomes with lysosomes in SH-SY5Y cells and induces Aβ deposition and cognitive dysfunction in STZ-induced diabetic rats. Behav. Brain Res. 2023, 442, 114286. [Google Scholar] [CrossRef]
- Biessels, G.J.; Nobili, F.; Teunissen, C.E.; Simó, R.; Scheltens, P. Understanding multifactorial brain changes in type 2 diabetes: A biomarker perspective. Lancet Neurol. 2020, 19, 699–710. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Sipols, A.; Kahn, S.E.; Lattemann, D.F.; Taborsky, G.J., Jr.; Bergman, R.N.; Woods, S.C.; Porte, D., Jr. Kinetics and specificity of insulin uptake from plasma into cerebrospinal fluid. Am. J. Physiol. 1990, 259, E378–E383. [Google Scholar] [CrossRef]
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516. [Google Scholar] [CrossRef]
- Ott, V.; Lehnert, H.; Staub, J.; Wönne, K.; Born, J.; Hallschmid, M. Central nervous insulin administration does not potentiate the acute glucoregulatory impact of concurrent mild hyperinsulinemia. Diabetes 2015, 64, 760–765. [Google Scholar] [CrossRef] [Green Version]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reger, M.A.; Watson, G.S.; Frey, W.H., 2nd; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. [Google Scholar] [CrossRef]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Benjanuwattra, J.; Apaijai, N.; Chunchai, T.; Kerdphoo, S.; Jaiwongkam, T.; Arunsak, B.; Wongsuchai, S.; Chattipakorn, N.; Chattipakorn, S.C. Metformin preferentially provides neuroprotection following cardiac ischemia/reperfusion in non-diabetic rats. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165893. [Google Scholar] [CrossRef] [PubMed]
- Khaleghi-Mehr, M.; Delshad, A.A.; Shafie-Damavandi, S.; Roghani, M. Metformin mitigates amyloid β1-40-induced cognitive decline via attenuation of oxidative/nitrosative stress and neuroinflammation. Metab. Brain Dis. 2023, 38, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Xu, J.; Li, B.; Ruan, Y.; Li, T.; Liu, J. Deciphering the Roles of Metformin in Alzheimer’s Disease: A Snapshot. Front. Pharmacol. 2022, 12, 728315. [Google Scholar] [CrossRef]
- Wu, C.Y.; Ouk, M.; Wong, Y.Y.; Anita, N.Z.; Edwards, J.D.; Yang, P.; Shah, B.R.; Herrmann, N.; Lanctôt, K.L.; Kapral, M.K.; et al. Relationships between memory decline and the use of metformin or DPP4 inhibitors in people with type 2 diabetes with normal cognition or Alzheimer’s disease, and the role APOE carrier status. Alzheimer’s Dement. 2020, 16, 1663–1673. [Google Scholar] [CrossRef]
- Bou Zerdan, M.; Hebbo, E.; Hijazi, A.; El Gemayel, M.; Nasr, J.; Nasr, D.; Yaghi, M.; Bouferraa, Y.; Nagarajan, A. The Gut Microbiome and Alzheimer’s Disease: A Growing Relationship. Curr. Alzheimer Res. 2022, 19, 808–818. [Google Scholar] [CrossRef]
- Cook, J.; Prinz, M. Regulation of microglial physiology by the microbiota. Gut Microbes 2022, 14, 2125739. [Google Scholar] [CrossRef]
- González-Arancibia, C.; Urrutia-Piñones, J.; Illanes-González, J.; Martinez-Pinto, J.; Sotomayor-Zárate, R.; Julio-Pieper, M.; Bravo, J.A. Do your gut microbes affect your brain dopamine? Psychopharmacology 2019, 236, 1611–1622. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, J.; Chen, Y. Regulation of Neurotransmitters by the Gut Microbiota and Effects on Cognition in Neurological Disorders. Nutrients 2021, 13, 2099. [Google Scholar] [CrossRef]
- Dicks, L.M.T. Gut Bacteria and Neurotransmitters. Microorganisms 2022, 10, 1838. [Google Scholar] [CrossRef]
- Choi, B.S.-Y.; Daoust, L.; Pilon, G.; Marette, A.; Tremblay, A. Potential therapeutic applications of the gut microbiome in obesity: From brain function to body detoxification. Int. J. Obes. 2020, 44, 1818–1831. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, Q.; Wang, Y.; Sun, A.; Lin, Y.; Jin, Y.; Li, X. Fecal microbiota transplantation from chronic unpredictable mild stress mice donors affects anxiety-like and depression-like behavior in recipient mice via the gut microbiota-inflammation-brain axis. Stress 2019, 22, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.F.; Zhu, Y.L.; Zhou, Z.L.; Jia, X.B.; Xu, Y.D.; Yang, Q.; Cui, C.; Shen, Y.Q. Neuroprotective Effects of Fecal Microbiota Transplantation on MPTP-Induced Parkinson’s Disease Mice: Gut Microbiota, Glial Reaction and TLR4/TNF-Alpha Signaling Pathway. Brain Behav. Immun. 2018, 70, 48–60. [Google Scholar] [CrossRef]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Jeon, S.H.; Ju, I.G.; Gee, M.S.; Do, J.; Oh, M.S.; Lee, J.K. Transplantation of gut microbiota derived from Alzheimer’s disease mouse model impairs memory function and neurogenesis in C57BL/6 mice. Brain Behav. Immun. 2021, 98, 357–365. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Li, X.H.; Liu, P.; Li, J.; Liu, L. The relationship between Alzheimer’s disease and intestinal microflora structure and inflammatory factors. Front. Aging Neurosci. 2022, 14, 972982. [Google Scholar] [CrossRef]
- Zhu, Z.; Ma, X.; Wu, J.; Xiao, Z.; Wu, W.; Ding, S.; Zheng, L.; Liang, X.; Luo, J.; Ding, D.; et al. Altered Gut Microbiota and Its Clinical Relevance in Mild Cognitive Impairment and Alzheimer’s Disease: Shanghai Aging Study and Shanghai Memory Study. Nutrients 2022, 14, 3959. [Google Scholar] [CrossRef]
- Borsom, E.M.; Conn, K.; Keefe, C.R.; Herman, C.; Orsini, G.M.; Hirsch, A.H.; Palma Avila, M.; Testo, G.; Jaramillo, S.A.; Bolyen, E.; et al. Predicting Neurodegenerative Disease Using Prepathology Gut Microbiota Composition: A Longitudinal Study in Mice Modeling Alzheimer’s Disease Pathologies. Microbiol. Spectr. 2023, 11, e0345822. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.L.; Heaver, S.L.; Walters, W.A.; Ley, R.E. Microbiome and metabolic disease: Revisiting the bacterial phylum Bacteroidetes. J. Mol. Med. 2017, 95, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhan, X.; Stamova, B.; Jin, L.W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [Green Version]
- Afzaal, M.; Saeed, F.; Hussain, M.; Ismail, Z.; Siddeeg, A.; Al-Farga, A.; Aljobair, M.O. Influence of encapsulation on the survival of probiotics in food matrix under simulated stress conditions. Saudi J. Biol. Sci. 2022, 29, 103394. [Google Scholar] [CrossRef] [PubMed]
- Pápai, G.; Torres-Maravilla, E.; Chain, F.; Varga-Visi, É.; Antal, O.; Naár, Z.; Bermúdez-Humarán, L.G.; Langella, P.; Martín, R. The Administration Matrix Modifies the Beneficial Properties of a Probiotic Mix of Bifidobacterium animalis subsp. lactis BB-12 and Lactobacillus acidophilus LA-5. Probiotics Antimicrob. Proteins 2021, 13, 484–494. [Google Scholar] [CrossRef]
- Lee, D.Y.; Shin, Y.J.; Kim, J.K.; Jang, H.M.; Joo, M.K.; Kim, D.H. Alleviation of cognitive impairment by gut microbiota lipopolysaccharide production-suppressing Lactobacillus plantarum and Bifidobacterium longum in mice. Food Funct. 2021, 12, 10750–10763. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, K.E.; Kim, J.K.; Kim, D.H. Suppression of gut dysbiosis by Bifidobacterium longum alleviates cognitive decline in 5XFAD transgenic and aged mice. Sci. Rep. 2019, 9, 11814. [Google Scholar] [CrossRef] [Green Version]
- Akbari, E.; Asemi, Z.; Daneshvar Kakhaki, R.; Bahmani, F.; Kouchaki, E.; Tamtaji, O.R.; Hamidi, G.A.; Salami, M. Effect of Probiotic Supplementation on Cognitive Function and Metabolic Status in Alzheimer’s Disease: A Randomized, Double-Blind and Controlled Trial. Front. Aging Neurosci. 2016, 8, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drljača, J.; Milošević, N.; Milanović, M.; Abenavoli, L.; Milić, N. When the microbiome helps the brain-current evidence. CNS Neurosci. Ther. 2023. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef] [PubMed]
- Villar-Gómez, N.; Ojeda-Hernandez, D.D.; López-Muguruza, E.; García-Flores, S.; Bonel-García, N.; Benito-Martín, M.S.; Selma-Calvo, B.; Canales-Aguirre, A.A.; Mateos-Díaz, J.C.; Montero-Escribano, P.; et al. Nose-to-Brain: The Next Step for Stem Cell and Biomaterial Therapy in Neurological Disorders. Cells 2022, 11, 3095. [Google Scholar] [CrossRef]
- Sánchez-Dengra, B.; González-Álvarez, I.; Bermejo, M.; González-Álvarez, M. Access to the CNS: Strategies to overcome the BBB. Int. J. Pharm. 2023, 636, 122759. [Google Scholar] [CrossRef] [PubMed]
- Ribovski, L.; Hamelmann, N.M.; Paulusse, J.M.J. Polymeric nanoparticles properties and brain delivery. Pharmaceutics 2021, 13, 2045. [Google Scholar] [CrossRef]
- Male, D.; Gromnicova, R. Nanocarriers for Delivery of Oligonucleotides to the CNS. Int. J. Mol. Sci. 2022, 23, 760. [Google Scholar] [CrossRef]
- Chakraborty, A.; Mohapatra, S.S.; Barik, S.; Roy, I.; Gupta, B.; Biswas, A. Impact of nanoparticles on amyloid β-induced Alzheimer’s disease, tuberculosis, leprosy and cancer: A systematic review. Biosci. Rep. 2023, 43, BSR20220324. [Google Scholar] [CrossRef]
- Zhong, X.; Na, Y.; Yin, S.; Yan, C.; Gu, J.; Zhang, N.; Geng, F. Cell Membrane Biomimetic Nanoparticles with Potential in Treatment of Alzheimer’s Disease. Molecules 2023, 28, 2336. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, H.; Yan, J.; Wei, Y.; Su, J. Engineered biomembrane-derived nanoparticles for nanoscale theranostics. Theranostics 2023, 13, 20–39. [Google Scholar] [CrossRef]
- Bayda, S.; Adeel, M.; Tuccinardi, T.; Cordani, M.; Rizzolio, F. The History of Nanoscience and Nanotechnology: From Chemical-Physical Applications to Nanomedicine. Molecules 2019, 25, 112. [Google Scholar] [CrossRef] [Green Version]
- Mendonça, M.C.P.; Sun, Y.; Cronin, M.F.; Lindsay, A.J.; Cryan, J.F.; O’Driscoll, C.M. Cyclodextrin-Based Nanoparticles for Delivery of Antisense Oligonucleotides Targeting Huntingtin. Pharmaceutics 2023, 15, 520. [Google Scholar] [CrossRef]
- He, L.; Huang, G.; Liu, H.; Sang, C.; Chen, T. Highly bioactive zeolitic imidazolate framework-8–capped nanotherapeutics for efficient reversal of reperfusion-induced injury in ischemic stroke. Sci. Adv. 2020, 6, eaay9751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhtar, A.; Andleeb, A.; Waris, T.S.; Bazzar, M.; Moradi, A.-R.; Awan, N.R.; Yar, M. Neurodegenerative diseases and effective drug delivery: A review of challenges and novel therapeutics. J. Control Release 2021, 330, 1152–1167. [Google Scholar] [CrossRef]
- Henriques, D.; Moreira, R.; Schwamborn, J.; Pereira de Almeida, L.; Mendonça, L.S. Successes and Hurdles in Stem Cells Application and Production for Brain Transplantation. Front. Neurosci. 2019, 13, 1194. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Jahan, S.; Singh, S.; Khanna, V.K.; Pant, A.B. Progress toward the development of in vitro model system for chemical-induced developmental neurotoxicity: Potential applicability of stem cells. Arch. Toxicol. 2015, 89, 265–267. [Google Scholar] [CrossRef]
- Monti, M.; Perotti, C.; Del Fante, C.; Cervio, M.; Redi, C.A.; Fondazione IRCCS Policlinico San Matteo, Pavia (Italia). Stem cells: Sources and therapies. Biol. Res. 2012, 45, 207–214. [Google Scholar] [CrossRef]
- Yamanaka, S. Pluripotent stem cell-based cell therapy-promise and challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Bhartiya, D. Are Mesenchymal Cells Indeed Pluripotent Stem Cells or Just Stromal Cells? OCT-4 and VSELs Biology Has Led to Better Understanding. Stem Cells Int. 2013, 2013, 547501. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Galiakberova, A.A.; Dashinimaev, E.B. Neural Stem Cells and Methods for Their Generation from Induced Pluripotent Stem Cells in vitro. Front. Cell Dev. Biol. 2020, 8, 815. [Google Scholar] [CrossRef]
- Zomer, H.D.; Vidane, A.S.; Gonçalves, N.N.; Ambrósio, C.E. Mesenchymal and induced pluripotent stem cells: General insights and clinical perspectives. Stem Cells Cloning 2015, 8, 125–134. [Google Scholar] [CrossRef]
- Noisa, P.; Raivio, T.; Cui, W. Neural Progenitor Cells Derived from Human Embryonic Stem Cells as an Origin of Dopaminergic Neurons. Stem Cells Int. 2015, 2015, 647437. [Google Scholar] [CrossRef] [Green Version]
- Mesulam, M.; Shaw, P.; Mash, D.; Weintraub, S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann. Neurol. 2004, 55, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, C.J.; Lyass, L.; Bhattacharyya, B.J.; Belmadani, A.; Miller, R.J.; Kessler, J.A. The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells 2011, 29, 802–811. [Google Scholar] [CrossRef] [Green Version]
- Yue, W.; Li, Y.; Zhang, T.; Jiang, M.; Qian, Y.; Zhang, M.; Sheng, N.; Feng, S.; Tang, K.; Yu, X.; et al. ESC-Derived Basal Forebrain Cholinergic Neurons Ameliorate the Cognitive Symptoms Associated with Alzheimer’s Disease in Mouse Models. Stem Cell Rep. 2015, 5, 776–790. [Google Scholar] [CrossRef] [Green Version]
- McGinley, L.M.; Kashlan, O.N.; Bruno, E.S.; Chen, K.S.; Hayes, J.M.; Kashlan, S.R.; Raykin, J.; Johe, K.; Murphy, G.G.; Feldman, E.L. Human neural stem cell transplantation improves cognition in a murine model of Alzheimer’s disease. Sci. Rep. 2018, 8, 14776. [Google Scholar] [CrossRef] [PubMed]
- McGinley, L.M.; Sims, E.; Lunn, J.S.; Kashlan, O.N.; Chen, K.S.; Bruno, E.S.; Pacut, C.M.; Hazel, T.; Johe, K.; Sakowski, S.A.; et al. Human Cortical Neural Stem Cells Expressing Insulin-Like Growth Factor-I: A Novel Cellular Therapy for Alzheimer’s Disease. Stem Cells Transl. Med. 2016, 5, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.A.; Yuan, C.X.; Liu, K.F.; Yang, Q.F.; Zhao, J.; Li, H.; Yang, Q.H.; Song, D.; Quan, Z.Z.; Qing, H. Neural stem cell transplantation alleviates functional cognitive deficits in a mouse model of tauopathy. Neural Regen. Res. 2022, 17, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Weick, J.P.; Liu, H.; Krencik, R.; Zhang, X.; Ma, L.; Zhou, G.M.; Ayala, M.; Zhang, S.C. Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol. 2013, 31, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Barker, R.A.; de Beaufort, I. Scientific and ethical issues related to stem cell research and interventions in neurodegenerative disorders of the brain. Prog. Neurobiol. 2013, 110, 63–73. [Google Scholar] [CrossRef]
- Li, J.Y.; Christophersen, N.S.; Hall, V.; Soulet, D.; Brundin, P. Critical issues of clinical human embryonic stem cell therapy for brain repair. Trends Neurosci. 2008, 31, 146–153. [Google Scholar] [CrossRef]
- Duan, Y.; Lyu, L.; Zhan, S. Stem Cell Therapy for Alzheimer’s Disease: A Scoping Review for 2017–2022. Biomedicines 2023, 11, 120. [Google Scholar] [CrossRef]
- Liu, X.Y.; Yang, L.P.; Zhao, L. Stem cell therapy for Alzheimer’s disease. World J. Stem Cells. 2020, 12, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, K.; Takeda, T.; Komori, N.; Tahara, K.; Yamagishi, H. Hypothesis: Intravenous administration of mesenchymal stem cells is effective in the treatment of Alzheimer’s disease. Med. Hypotheses 2021, 150, 110572. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, H.; Shigematsu, K. Perspectives on Stem Cell-Based Regenerative Medicine with a Particular Emphasis on Mesenchymal Stem Cell Therapy. JMA J. 2022, 5, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Ahani-Nahayati, M.; Shariati, A.; Mahmoodi, M.; Olegovna Zekiy, A.; Javidi, K.; Shamlou, S.; Mousakhani, A.; Zamani, M.; Hassanzadeh, A. Stem cell in neurodegenerative disorders; an emerging strategy. Int. J. Dev. Neurosci. 2021, 81, 291–311. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, W.; Wang, X.; Yang, J.; Cui, Y.; Song, H.; Li, W.; Li, W.; Wu, L.; Du, Y.; et al. A passage-dependent network for estimating the in vitro senescence of mesenchymal stromal/stem cells using microarray, bulk and single cell RNA sequencing. Front. Cell Dev. Biol. 2023, 7, 998666. [Google Scholar] [CrossRef]
- Duncan, T.; Valenzuela, M. Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res. Ther. 2017, 8, 111. [Google Scholar] [CrossRef]
- Joyce, N.; Annett, G.; Wirthlin, L.; Olson, S.; Bauer, G.; Nolta, J.A. Mesenchymal stem cells for the treatment of neurodegenerative disease. Regen. Med. 2010, 5, 933–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naaldijk, Y.; Jäger, C.; Fabian, C.; Leovsky, C.; Blüher, A.; Rudolph, L.; Hinze, A.; Stolzing, A. Effect of systemic transplantation of bone marrow-derived mesenchymal stem cells on neuropathology markers in APP/PS1 Alzheimer mice. Neuropathol. Appl. Neurobiol. 2017, 43, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Park, B.N.; Kim, J.H.; Lim, T.S.; Park, S.H.; Kim, T.G.; Yoon, B.S.; Son, K.S.; Yoon, J.K.; An, Y.S. Therapeutic effect of mesenchymal stem cells in an animal model of Alzheimer’s disease evaluated by β-amyloid positron emission tomography imaging. Aust. N. Z. J. Psychiatry 2020, 54, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xie, Z.; Bi, J.; Zhu, Z. Anti-inflammatory effects of bone marrow mesenchymal stem cells on mice with Alzheimer’s disease. Exp. Ther. Med. 2018, 16, 5015–5020. [Google Scholar] [CrossRef] [Green Version]
- Crigler, L.; Robey, R.C.; Asawachaicharn, A.; Gaupp, D.; Phinney, D.G. Human mesenchymal stem cell subpopulations express a variety of neuro-regulatory molecules and promote neuronal cell survival and neuritogenesis. Exp. Neurol. 2006, 198, 54–64. [Google Scholar] [CrossRef]
- Lee, J.K.; Jin, H.K.; Endo, S.; Schuchman, E.H.; Carter, J.E.; Bae, J.S. Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-beta deposition and rescues memory deficits in Alzheimer’s disease mice by modulation of immune responses. Stem Cells 2010, 28, 329–343. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, J.K.; Lee, H.; Carter, J.E.; Chang, J.W.; Oh, W.; Yang, Y.S.; Suh, J.G.; Lee, B.H.; Jin, H.K.; et al. Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 2012, 33, 588–602. [Google Scholar] [CrossRef]
- Boutajangout, A.; Noorwali, A.; Atta, H.; Wisniewski, T. Human Umbilical Cord Stem Cell Xenografts Improve Cognitive Decline and Reduce the Amyloid Burden in a Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 104–111. [Google Scholar] [CrossRef]
- Kim, H.J.; Seo, S.W.; Chang, J.W.; Lee, J.I.; Kim, C.H.; Chin, J.; Choi, S.J.; Kwon, H.; Yun, H.J.; Lee, J.M.; et al. Stereotactic brain injection of human umbilical cord blood mesenchymal stem cells in patients with Alzheimer’s disease dementia: A phase 1 clinical trial. Alzheimer’s Dement 2015, 1, 95–102. [Google Scholar] [CrossRef]
- Kim, H.J.; Cho, K.R.; Jang, H.; Lee, N.K.; Jung, Y.H.; Kim, J.P.; Lee, J.I.; Chang, J.W.; Park, S.; Kim, S.T.; et al. Intracerebroventricular injection of human umbilical cord blood mesenchymal stem cells in patients with Alzheimer’s disease dementia: A phase I clinical trial. Alzheimers Res. Ther. 2021, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Myeong, S.H.; Kim, H.; Lee, N.K.; Hwang, J.W.; Kim, H.J.; Jang, H.; Choi, S.J.; Na, D.L. Intracerebroventricular Administration of Human Umbilical Cord Blood-Derived Mesenchymal Stem Cells Induces Transient Inflammation in a Transgenic Mouse Model and Patients with Alzheimer’s Disease. Biomedicines 2022, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren Bendtsen, K.M.; Hall, V.J. The Breakthroughs and Caveats of Using Human Pluripotent Stem Cells in Modeling Alzheimer’s Disease. Cells 2023, 12, 420. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Jimenez, F.J.; Ureña-Peralta, J.; Jendelova, P.; Erceg, S. Alzheimer’s disease and synapse Loss: What can we learn from induced pluripotent stem Cells? J. Adv. Res. 2023. [Google Scholar] [CrossRef]
- Tcw, J. Human iPSC application in Alzheimer’s disease and Tau-related neurodegenerative diseases. Neurosci. Lett. 2019, 699, 31–40. [Google Scholar] [CrossRef]
- Kim, J.Y.; Nam, Y.; Rim, Y.A.; Ju, J.H. Review of the Current Trends in Clinical Trials Involving Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2022, 18, 142–154. [Google Scholar] [CrossRef]
- Takahashi, J. iPS cell-based therapy for Parkinson’s disease: A Kyoto trial. Regen. Ther. 2020, 13, 18–22. [Google Scholar] [CrossRef]
- Takahashi, J. Clinical Trial for Parkinson’s Disease Gets a Green Light in the US. Cell Stem Cell 2021, 28, 182–183. [Google Scholar] [CrossRef]
- Yefroyev, D.A.; Jin, S. Induced Pluripotent Stem Cells for Treatment of Alzheimer’s and Parkinson’s Diseases. Biomedicines 2022, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, M.; Fomenko, A.; Lozano, A.M.; Kiening, K.L. Cellular, molecular, and clinical mechanisms of action of deep brain stimulation-a systematic review on established indications and outlook on future developments. EMBO Mol. Med. 2019, 11, e9575. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Lee, J.; Liu, C.Y.; Lozano, A.M.; Lee, D.J. Deep Brain Stimulation for Alzheimer’s Disease: Tackling Circuit Dysfunction. Neuromodulation 2021, 24, 171–186. [Google Scholar] [CrossRef]
- Xu, J.; Huang, T.; Dana, A. Deep brain stimulation of the subthalamic nucleus to improve symptoms and cognitive functions in patients with refractory obsessive-compulsive disorder: A longitudinal study. Neurol. Sci. 2023. [Google Scholar] [CrossRef]
- Boogers, A.; Peeters, J.; Van Bogaert, T.; Asamoah, B.; De Vloo, P.; Vandenberghe, W.; Nuttin, B.; Mc Laughlin, M. Anodic and symmetric biphasic pulses enlarge the therapeutic window in deep brain stimulation for essential tremor. Brain Stimul. 2022, 15, 286–290. [Google Scholar] [CrossRef]
- Peeters, J.; Boogers, A.; Van Bogaert, T.; Davidoff, H.; Gransier, R.; Wouters, J.; Nuttin, B.; Mc Laughlin, M. Electrophysiologic Evidence That Directional Deep Brain Stimulation Activates Distinct Neural Circuits in Patients with Parkinson Disease. Neuromodulation 2023, 26, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Zhen, J.; Qian, Y.; Fu, J.; Su, R.; An, H.; Wang, W.; Zheng, Y.; Wang, X. Deep Brain Magnetic Stimulation Promotes Neurogenesis and Restores Cholinergic Activity in a Transgenic Mouse Model of Alzheimer’s Disease. Front. Neural. Circuits 2017, 11, 48. [Google Scholar] [CrossRef]
- Jakobs, M.; Lee, D.J.; Lozano, A.M. Modifying the progression of Alzheimer’s and Parkinson’s disease with deep brain stimulation. Neuropharmacology 2020, 171, 107860. [Google Scholar] [CrossRef]
- Puig-Parnau, I.; Garcia-Brito, S.; Vila-Soles, L.; Riberas, A.; Aldavert-Vera, L.; Segura-Torres, P.; Kádár, E.; Huguet, G. Intracranial Self-stimulation of the Medial Forebrain Bundle Ameliorates Memory Disturbances and Pathological Hallmarks in an Alzheimer’s Disease Model by Intracerebral Administration of Amyloid-β in Rats. Neuroscience 2023, 512, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yuan, T.S.; Chen, Y.C.; Guo, P.; Lian, T.H.; Liu, Y.Y.; Liu, W.; Bai, Y.T.; Zhang, Q.; Zhang, W.; et al. Deep brain stimulation of the nucleus basalis of Meynert modulates hippocampal-frontoparietal networks in patients with advanced Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 51. [Google Scholar] [CrossRef]
- Cheyuo, C.; Germann, J.; Yamamoto, K.; Vetkas, A.; Loh, A.; Sarica, C.; Milano, V.; Zemmar, A.; Flouty, O.; Harmsen, I.E.; et al. Connectomic neuromodulation for Alzheimer’s disease: A systematic review and meta-analysis of invasive and non-invasive techniques. Transl. Psychiatry 2022, 12, 490. [Google Scholar] [CrossRef] [PubMed]
- Leoutsakos, J.S.; Yan, H.; Anderson, W.S.; Asaad, W.F.; Baltuch, G.; Burke, A.; Chakravarty, M.M.; Drake, K.E.; Foote, K.D.; Fosdick, L.; et al. Deep Brain Stimulation Targeting the Fornix for Mild Alzheimer Dementia (the ADvance Trial): A Two Year Follow-up Including Results of Delayed Activation. J. Alzheimer’s Dis. 2018, 64, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Scharre, D.W.; Weichart, E.; Nielson, D.; Zhang, J.; Agrawal, P.; Sederberg, P.B.; Knopp, M.V.; Rezai, A.R. Alzheimer’s Disease Neuroimaging Initiative Deep Brain Stimulation of Frontal Lobe Networks to Treat Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shu, K.; Geng, Y.; Cai, C.; Kang, H. Deep brain stimulation of fornix in Alzheimer’s disease: From basic research to clinical practice. Eur. J. Clin. Invest. 2023. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Sun, Y.; Tian, X.; Zheng, X.; Wang, X.; Li, W.; Wu, X.; Shu, B.; Hou, W. Deep Brain Stimulation for Alzheimer’s Disease: Stimulation Parameters and Potential Mechanisms of Action. Front. Aging Neurosci. 2021, 13, 619543. [Google Scholar] [CrossRef] [PubMed]
- Alves, P.N.; Foulon, C.; Karolis, V.; Bzdok, D.; Margulies, D.S.; Volle, E.; Thiebaut de Schotten, M. An improved neuroanatomical model of the default-mode network reconciles previous neuroimaging and neuropathological findings. Commun. Biol. 2019, 2, 370. [Google Scholar] [CrossRef] [Green Version]
- Hamani, C.; McAndrews, M.P.; Cohn, M.; Oh, M.; Zumsteg, D.; Shapiro, C.M.; Wennberg, R.A.; Lozano, A.M. Memory enhancement induced by hypothalamic/fornix deep brain stimulation. Ann. Neurol. 2008, 63, 119–123. [Google Scholar] [CrossRef]
- Sankar, T.; Chakravarty, M.M.; Bescos, A.; Lara, M.; Obuchi, T.; Laxton, A.W.; McAndrews, M.P.; Tang-Wai, D.F.; Workman, C.I.; Smith, G.S.; et al. Deep Brain Stimulation Influences Brain Structure in Alzheimer’s Disease. Brain Stimul. 2015, 8, 645–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, A.M.; Fosdick, L.; Chakravarty, M.M.; Leoutsakos, J.M.; Munro, C.; Oh, E.; Drake, K.E.; Lyman, C.H.; Rosenberg, P.B.; Anderson, W.S.; et al. A phase II study of fornix deep brain stimulation in mild Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 54, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leplus, A.; Lauritzen, I.; Melon, C.; Kerkerian-Le Goff, L.; Fontaine, D.; Checler, F. Chronic fornix deep brain stimulation in a transgenic Alzheimer’s rat model reduces amyloid burden, inflammation, and neuronal loss. Brain Struct Funct. 2019, 224, 363–372. [Google Scholar] [CrossRef]
- Gallino, D.; Devenyi, G.A.; Germann, J.; Guma, E.; Anastassiadis, C.; Chakravarty, M.M. Longitudinal assessment of the neuroanatomical consequences of deep brain stimulation: Application of fornical DBS in an Alzheimer’s mouse model. Brain Res. 2019, 1715, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Ríos, A.S.; Oxenford, S.; Neudorfer, C.; Butenko, K.; Li, N.; Rajamani, N.; Boutet, A.; Elias, G.J.B.; Germann, J.; Loh, A.; et al. Optimal deep brain stimulation sites and networks for stimulation of the fornix in Alzheimer’s disease. Nat. Commun. 2022, 13, 7707. [Google Scholar] [CrossRef]
- Aldehri, M.; Temel, Y.; Alnaami, I.; Jahanshahi, A.; Hescham, S. Deep brain stimulation for Alzheimer’s Disease: An update. Surg. Neurol. Int. 2018, 9, 58. [Google Scholar] [CrossRef]
- Yu, D.; Yan, H.; Zhou, J.; Yang, X.; Lu, Y.; Han, Y. A circuit view of deep brain stimulation in Alzheimer’s disease and the possible mechanisms. Mol. Neurodegener. 2019, 14, 33. [Google Scholar] [CrossRef] [Green Version]
- Zarzycki, M.Z.; Domitrz, I. Stimulation-induced side effects after deep brain stimulation—A systematic review. Acta Neuropsychiatr. 2020, 32, 57–64. [Google Scholar] [CrossRef]
- Kantzanou, M.; Korfias, S.; Panourias, I.; Sakas, D.E.; Karalexi, M.A. Deep Brain Stimulation-Related Surgical Site Infections: A Systematic Review and Meta-Analysis. Neuromodulation 2021, 24, 197–211. [Google Scholar] [CrossRef]
- Bittlinger, M.; Müller, S. Opening the debate on deep brain stimulation for Alzheimer disease—A critical evaluation of rationale, shortcomings, and ethical justification. BMC Med. Ethics 2018, 19, 41. [Google Scholar] [CrossRef] [Green Version]
- Hunt, N.J.; Wahl, D.; Westwood, L.J.; Lockwood, G.P.; Le Couteur, D.G.; Cogger, V.C. Targeting the liver in dementia and cognitive impairment: Dietary macronutrients and diabetic therapeutics. Adv. Drug Deliv. Rev. 2022, 190, 114537. [Google Scholar] [CrossRef]
- Ribaric, S. Physical exercise, a potential non-pharmacological intervention for attenuating neuroinflammation and cognitive decline in Alzheimer’s disease patients. Int. J. Mol. Sci. 2022, 23, 3245. [Google Scholar] [CrossRef]
- Devanand, D.P.; Masurkar, A.V.; Wisniewski, T. Vigorous, regular physical exercise may slow disease progression in Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Antioxidant vitamin intake and risk of Alzheimer disease. Arch. Neurol. 2003, 60, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Schupf, N.; Cruz, E.; Stern, Y.; Mayeux, R.P.; Gu, Y. Association Between Mediterranean Diet and Functional Status in Older Adults: A Longitudinal Study Based on the Washington Heights-Inwood Columbia Aging Project. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Trichopoulou, A.; Martínez-González, M.A.; Tong, T.Y.; Forouhi, N.G.; Khandelwal, S.; Prabhakaran, D.; Mozaffarian, D.; de Lorgeril, M. Definitions and potential health benefits of the Mediterranean diet: Views from experts around the world. BMC Med. 2014, 12, 112. [Google Scholar] [CrossRef] [Green Version]
- Filippou, C.D.; Tsioufis, C.P.; Thomopoulos, C.G.; Mihas, C.C.; Dimitriadis, K.S.; Sotiropoulou, L.I.; Chrysochoou, C.A.; Nihoyannopoulos, P.I.; Tousoulis, D.M. Dietary Approaches to Stop Hypertension (DASH) Diet and Blood Pressure Reduction in Adults with and without Hypertension: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Adv. Nutr. 2020, 11, 1150–1160. [Google Scholar] [CrossRef]
- Black, L.J.; Baker, K.; Ponsonby, A.L.; van der Mei, I.; Lucas, R.M.; Pereira, G.; Ausimmune Investigator Group. A Higher Mediterranean Diet Score, Including Unprocessed Red Meat, Is Associated with Reduced Risk of Central Nervous System Demyelination in a Case-Control Study of Australian Adults. J. Nutr. 2019, 149, 1385–1392. [Google Scholar] [CrossRef]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Bennett, D.A.; Aggarwal, N.T. MIND Diet associated with reduced incidence of Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Scarmeas, N.; Stern, Y.; Tang, M.X.; Mayeux, R.; Luchsinger, J.A. Mediterranean diet and risk for Alzheimer’s disease. Ann. Neurol. 2006, 59, 912–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarmeas, N.; Stern, Y.; Mayeux, R.; Manly, J.J.; Schupf, N.; Luchsinger, J.A. Mediterranean diet and mild cognitive impairment. Arch. Neurol. 2009, 66, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangney, C.C.; Kwasny, M.J.; Li, H.; Wilson, R.S.; Evans, D.A.; Morris, M.C. Adherence to a Mediterranean-type dietary pattern and cognitive decline in a community population. Am. J. Clin. Nutr. 2011, 93, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaynor, A.M.; Varangis, E.; Song, S.; Gazes, Y.; Noofoory, D.; Babukutty, R.S.; Habeck, C.; Stern, Y.; Gu, Y. Diet moderates the effect of resting state functional connectivity on cognitive function. Sci. Rep. 2022, 12, 16080. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Beck, T.; Bennett, D.A.; Schneider, J.A.; Hayden, K.M.; Shadyab, A.H.; Rajan, K.B.; Morris, M.C.; Cornelis, M.C. Adherence to MIND Diet, Genetic Susceptibility, and Incident Dementia in Three US Cohorts. Nutrients 2022, 14, 2759. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, B.; Trifan, G.; Isasi, C.R.; Lipton, R.B.; Sotres-Alvarez, D.; Cai, J.; Tarraf, W.; Stickel, A.; Mattei, J.; Talavera, G.A.; et al. Association of Mediterranean Diet with Cognitive Decline Among Diverse Hispanic or Latino Adults From the Hispanic Community Health Study/Study of Latinos. JAMA Netw. Open 2022, 5, e2221982. [Google Scholar] [CrossRef]
- Agarwal, P.; Leurgans, S.E.; Agrawal, S.; Aggarwal, N.; Cherian, L.J.; James, B.D.; Dhana, K.; Barnes, L.L.; Bennett, D.A.; Schneider, J.A. Association of Mediterranean-DASH Intervention for Neurodegenerative Delay and Mediterranean Diets with Alzheimer Disease Pathology. Neurology 2023, 100, e2259–e2268. [Google Scholar] [CrossRef]
- Song, S.; Gaynor, A.M.; Cruz, E.; Lee, S.; Gazes, Y.; Habeck, C.; Stern, Y.; Gu, Y. Mediterranean Diet and White Matter Hyperintensity Change over Time in Cognitively Intact Adults. Nutrients 2022, 14, 3664. [Google Scholar] [CrossRef]
- Gauci, S.; Young, L.M.; Arnoldy, L.; Lassemillante, A.C.; Scholey, A.; Pipingas, A. Dietary patterns in middle age: Effects on concurrent neurocognition and risk of age-related cognitive decline. Nutr. Rev. 2022, 80, 1129–1159. [Google Scholar] [CrossRef]
- Chen, H.; Dhana, K.; Huang, Y.; Huang, L.; Tao, Y.; Liu, X.; Melo van Lent, D.; Zheng, Y.; Ascherio, A.; Willett, W.; et al. Association of the Mediterranean Dietary Approaches to Stop Hypertension Intervention for Neurodegenerative Delay (MIND) Diet With the Risk of Dementia. JAMA Psychiatry 2023, 80, 630–638. [Google Scholar] [CrossRef]
- Glans, I.; Sonestedt, E.; Nägga, K.; Gustavsson, A.M.; González-Padilla, E.; Borne, Y.; Stomrud, E.; Melander, O.; Nilsson, P.; Palmqvist, S.; et al. Association Between Dietary Habits in Midlife with Dementia Incidence Over a 20-Year Period. Neurology 2022, 100, e28–e37. [Google Scholar] [CrossRef] [PubMed]
- Wesselman, L.M.P.; van Lent, D.M.; Schröder, A.; van de Rest, O.; Peters, O.; Menne, F.; Fuentes, M.; Priller, J.; Spruth, E.J.; Altenstein, S.; et al. Dietary patterns are related to cognitive functioning in elderly enriched with individuals at increased risk for Alzheimer’s disease. Eur. J. Nutr. 2021, 60, 849–860. [Google Scholar] [CrossRef]
- Ballarini, T.; Melo van Lent, D.; Brunner, J.; Schröder, A.; Wolfsgruber, S.; Altenstein, S.; Brosseron, F.; Buerger, K.; Dechent, P.; Dobisch, L.; et al. Mediterranean Diet, Alzheimer Disease Biomarkers and Brain Atrophy in Old Age. Neurology 2021, 96, e2920–e2932. [Google Scholar] [CrossRef]
- Gregory, S.; Ritchie, C.W.; Ritchie, K.; Shannon, O.; Stevenson, E.J.; Muniz-Terrera, G. Mediterranean diet score is associated with greater allocentric processing in the EPAD LCS cohort: A comparative analysis by biogeographical region. Front. Aging 2022, 3, 1012598. [Google Scholar] [CrossRef] [PubMed]
- Mohan, D.; Yap, K.H.; Reidpath, D.; Soh, Y.C.; McGrattan, A.; Stephan, B.C.M.; Robinson, L.; Chaiyakunapruk, N.; Siervo, M.; DePEC team. Link Between Dietary Sodium Intake, Cognitive Function, and Dementia Risk in Middle-Aged and Older Adults: A Systematic Review. J. Alzheimers Dis. 2020, 76, 1347–1373. [Google Scholar] [CrossRef] [PubMed]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Freeman, L.R.; Haley-Zitlin, V.; Rosenberger, D.S.; Granholm, A.C. Damaging effects of a high-fat diet to the brain and cognition: A review of proposed mechanisms. Nutr. Neurosci. 2014, 17, 241–251. [Google Scholar] [CrossRef]
- Solfrizzi, V.; Custodero, C.; Lozupone, M.; Imbimbo, B.P.; Valiani, V.; Agosti, P.; Schilardi, A.; D’Introno, A.; La Montagna, M.; Calvani, M.; et al. Relationships of dietary patterns, foods, and micro- and macronutrients with Alzheimer’s disease and late-life cognitive disorders: A systematic review. J. Alzheimers Dis. 2017, 59, 815–849. [Google Scholar] [CrossRef] [Green Version]
- Gardener, S.L.; Rainey-Smith, S.R.; Barnes, M.B.; Sohrabi, H.R.; Weinborn, M.; Lim, Y.Y.; Harrington, K.; Taddei, K.; Gu, Y.; Rembach, A.; et al. Dietary patterns and cognitive decline in an Australian study of ageing. Mol. Psychiatry 2015, 20, 860–866. [Google Scholar] [CrossRef]
- Grant, W.B. Using Multicountry Ecological and Observational Studies to Determine Dietary Risk Factors for Alzheimer’s Disease. J. Am. Coll. Nutr. 2016, 35, 476–489. [Google Scholar] [CrossRef]
- Guillemot-Legris, O.; Muccioli, G.G. Obesity-induced Neuroinflammation: Beyond the hypothalamus. Trends Neurosci. 2017, 40, 237–253. [Google Scholar] [CrossRef]
- Pasinetti, G.M.; Eberstein, J.A. Metabolic syndrome and the role of dietary lifestyles in Alzheimer’s disease. J. Neurochem. 2008, 106, 1503–1514. [Google Scholar] [CrossRef] [Green Version]
- Więckowska-Gacek, A.; Mietelska-Porowska, A.; Wydrych, M.; Wojda, U. Western diet as a trigger of Alzheimer’s disease: From metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration. Ageing Res. Rev. 2021, 70, 101397. [Google Scholar] [CrossRef] [PubMed]
- Koerich, S.; Parreira, G.M.; de Almeida, D.L.; Vieira, R.P.; de Oliveira, A.C.P. Receptors for Advanced Glycation End Products (RAGE): Promising Targets Aiming at the Treatment of Neurodegenerative Conditions. Curr. Neuropharmacol. 2023, 21, 219–234. [Google Scholar] [CrossRef]
- Takeuchi, M.; Sato, T.; Takino, J.; Kobayashi, Y.; Furuno, S.; Kikuchi, S.; Yamagishi, S. Diagnostic utility of serum or cerebrospinal fluid levels of toxic advanced glycation end-products (TAGE) in early detection of Alzheimer’s disease. Med. Hypotheses 2007, 69, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef]
- Lotan, R.; Ganmore, I.; Livny, A.; Itzhaki, N.; Waserman, M.; Shelly, S.; Zacharia, M.; Moshier, E.; Uribarri, J.; Beisswenger, P.; et al. Effect of Advanced Glycation End Products on Cognition in Older Adults with Type 2 Diabetes: Results from a Pilot Clinical Trial. J. Alzheimers Dis. 2021, 82, 1785–1795. [Google Scholar] [CrossRef]
- Stecker, M.; Stecker, M.; Reiss, A.B.; Kasselman, L. Dementia and Diet, Methodological and Statistical Issues: A Pilot Study. Front. Aging Neurosci. 2022, 14, 606424. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, T.S. Calorie restriction or dietary restriction: How far they can protect the brain against neurodegenerative diseases? Neural Regen. Res. 2022, 17, 1640–1644. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, V.E.; Herrera, P.F.; Laura, R. Effect of nutrition on neurodegenerative diseases. A systematic review. Nutr. Neurosci. 2021, 24, 810–834. [Google Scholar] [CrossRef]
- Anastasiou, C.A.; Yannakoulia, M.; Kosmidis, M.H.; Dardiotis, E.; Hadjigeorgiou, G.M.; Sakka, P.; Arampatzi, X.; Bougea, A.; Labropoulos, I.; Scarmeas, N. Mediterranean diet and cognitive health: Initial results from the hellenic longitudinal investigation of ageing and diet. PLoS ONE 2017, 12, e0182048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, D.; Coogan, S.C.; Solon-Biet, S.M.; de Cabo, R.; Haran, J.B.; Raubenheimer, D.; Cogger, V.C.; Mattson, M.P.; Simpson, S.J.; Le Couteur, D.G. Cognitive and behavioral evaluation of nutritional interventions in rodent models of brain aging and dementia. Clin. Interv. Aging 2017, 12, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Gültekin, F.; Nazıroğlu, M.; Savaş, H.B.; Çiğ, B. Calorie restriction protects against apoptosis, mitochondrial oxidative stress and increased calcium signaling through inhibition of TRPV1 channel in the hippocampus and dorsal root ganglion of rats. Metab. Brain Dis. 2018, 33, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Halagappa, V.K.; Guo, Z.; Pearson, M.; Matsuoka, Y.; Cutler, R.G.; Laferla, F.M.; Mattson, M.P. Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Dis. 2007, 26, 212–220. [Google Scholar] [CrossRef]
- Witte, A.V.; Fobker, M.; Gellner, R.; Knecht, S.; Floel, A. Caloric restriction improves memory in elderly humans. Proc. Natl. Acad. Sci. USA 2009, 106, 1255–1260. [Google Scholar] [CrossRef] [Green Version]
- Bayer-Carter, J.L.; Green, P.S.; Montine, T.J.; VanFossen, B.; Baker, L.D.; Watson, G.S.; Bonner, L.M.; Callaghan, M.; Leverenz, J.B.; Walter, B.K.; et al. Diet intervention and cerebrospinal fluid biomarkers in amnestic mild cognitive impairment. Arch. Neurol. 2011, 68, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Wahl, D.; Cogger, V.C.; Solon-Biet, S.M.; Waern, R.V.; Gokarn, R.; Pulpitel, T.; Cabo, R.d.; Mattson, M.P.; Raubenheimer, D.; Simpson, S.J.; et al. Nutritional strategies to optimise cognitive function in the aging brain. Ageing Res. Rev. 2016, 31, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, A.; Padinjakara, N.; Lautenschlager, N.T. Effects of intermittent fasting on cognitive health and Alzheimer’s disease. Nutr. Rev. 2023. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Dursun, E.; Féron, F.; Gezen-Ak, D.; Kalueff, A.V.; Littlejohns, T.; Llewellyn, D.J.; Millet, P.; Scott, T.; Tucker, K.L.; et al. Vitamin D and cognition in older adults`: Updated international recommendations. J. Intern. Med. 2015, 277, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Nagel, G.; Herbolsheimer, F.; Riepe, M.; Nikolaus, T.; Denkinger, M.D.; Peter, R.; Weinmayr, G.; Rothenbacher, D.; Koenig, W.; Ludolph, A.C.; et al. Serum vitamin D concentrations and cognitive function in a population-based study among older adults in South Germany. J. Alzheimers Dis. 2015, 45, 1119–1126. [Google Scholar] [CrossRef]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the vitamin D receptor and 1α-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Garcion, E.; Wion-Barbot, N.; Montero-Menei, C.N.; Berger, F.; Wion, D. New clues about vitamin D functions in the nervous system. Trends Endocrinol. Metab. 2002, 13, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Uthaiah, C.A.; Beeraka, N.M.; Rajalakshmi, R.; Ramya, C.M.; Madhunapantula, S.V. Role of Neural Stem Cells and Vitamin D Receptor (VDR)-Mediated Cellular Signaling in the Mitigation of Neurological Diseases. Mol. Neurobiol. 2022, 59, 4065–4105. [Google Scholar] [CrossRef]
- Littlejohns, T.J.; Henley, W.E.; Lang, I.A.; Annweiler, C.; Beauchet, O.; Chaves, P.H.; Fried, L.; Kestenbaum, B.R.; Kuller, L.H.; Langa, K.M.; et al. Vitamin D and the risk of dementia and Alzheimer disease. Neurology 2014, 83, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Meng, L.; Wang, Z.; Ming, Y.C.; Shen, L.; Ji, H.F. Are micronutrient levels and supplements causally associated with the risk of Alzheimer’s disease? A two-sample Mendelian randomization analysis. Food Funct. 2022, 13, 6665–6673. [Google Scholar] [CrossRef]
- Balion, C.; Griffith, L.E.; Strifler, L.; Henderson, M.; Patterson, C.; Heckman, G.; Llewellyn, D.J.; Raina, P. Vitamin D, cognition, and dementia: A systematic review and meta-analysis. Neurology 2012, 79, 1397–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etgen, T.; Sander, D.; Bickel, H.; Sander, K.; Forstl, H. Vitamin D deficiency, cognitive impairment and dementia: A systematic review and meta-analysis. Dement. Geriatr. Cogn. Disord. 2012, 33, 297–305. [Google Scholar] [CrossRef]
- Kalra, A.; Teixeira, A.L.; Diniz, B.S. Association of Vitamin D Levels with Incident All-Cause Dementia in Longitudinal Observational Studies: A Systematic Review and Meta-analysis. J. Prev. Alzheimers Dis. 2020, 7, 14–20. [Google Scholar] [CrossRef]
- Chai, B.; Gao, F.; Wu, R.; Dong, T.; Gu, C.; Lin, Q.; Zhang, Y. Vitamin D deficiency as a risk factor for dementia and Alzheimer’s disease: An updated meta-analysis. BMC Neurol. 2019, 19, 284. [Google Scholar] [CrossRef]
- Jayedi, A.; Rashidy-Pour, A.; Shab-Bidar, S. Vitamin D status and risk of dementia and Alzheimer’s disease: A meta-analysis of dose-response. Nutr. Neurosci. 2019, 22, 750–759. [Google Scholar] [CrossRef]
- Du, Y.; Liang, F.; Zhang, L.; Liu, J.; Dou, H. Vitamin D Supplement for Prevention of Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Am. J. Ther. 2020, 28, e638–e648. [Google Scholar] [CrossRef]
- Bischoff-Ferrari, H.A.; Vellas, B.; Rizzoli, R.; Kressig, R.W.; da Silva, J.A.P.; Blauth, M.; Felson, D.T.; McCloskey, E.V.; Watzl, B.; Hofbauer, L.C.; et al. Effect of Vitamin D Supplementation, Omega-3 Fatty Acid Supplementation, or a Strength-Training Exercise Program on Clinical Outcomes in Older Adults: The DO-HEALTH Randomized Clinical Trial. JAMA. 2020, 324, 1855–1868. [Google Scholar] [CrossRef]
- Aspell, N.; Lawlor, B.; O’Sullivan, M. Is there a role for vitamin D in supporting cognitive function as we age? Proc. Nutr. Soc. 2018, 77, 124–134. [Google Scholar] [CrossRef]
- Fan, Y.G.; Pang, Z.Q.; Wu, T.Y.; Zhang, Y.H.; Xuan, W.Q.; Wang, Z.; Yu, X.; Li, Y.C.; Guo, C.; Wang, Z.Y. Vitamin D deficiency exacerbates Alzheimer-like pathologies by reducing antioxidant capacity. Free Radic. Biol. Med. 2020, 161, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Mehrabadi, S.; Sadr, S.S. Administration of Vitamin D3 and E supplements reduces neuronal loss and oxidative stress in a model of rats with Alzheimer’s disease. Neurol. Res. 2020, 42, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Morello, M.; Pieri, M.; Zenobi, R.; Talamo, A.; Stephan, D.; Landel, V.; Féron, F.; Millet, P. The Influence of Vitamin D on Neurodegeneration and Neurological Disorders: A Rationale for its Physio-pathological Actions. Curr. Pharm. Des. 2020, 26, 2475–2491. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C. Vitamin D in dementia prevention. Ann. N. Y. Acad. Sci. 2016, 1367, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, M.O.W.P.; Thiel, A.; Lauer, A.A.; Winkler, J.; Lehmann, J.; Regner, L.; Nelke, C.; Janitschke, D.; Benoist, C.; Streidenberger, O.; et al. Vitamin D and Its Analogues Decrease Amyloid- β (Aβ) Formation and Increase Aβ-Degradation. Int. J. Mol. Sci. 2017, 18, 2764. [Google Scholar] [CrossRef] [Green Version]
- Durk, M.R.; Han, K.; Chow, E.C.; Ahrens, R.; Henderson, J.T.; Fraser, P.E.; Pang, K.S. 1alpha,25-Dihydroxyvitamin D3 reduces cerebral amyloid-beta accumulation and improves cognition in mouse models of Alzheimer’s disease. J. Neurosci. 2014, 34, 7091–7101. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Gattoni-Celli, M.; Zhu, H.; Bhat, N.R.; Sambamurti, K.; Gattoni-Celli, S.; Kindy, M.S. Vitamin D3-enriched diet correlates with a decrease of amyloid plaques in the brain of AβPP transgenic mice. J. Alzheimers Dis. 2011, 25, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Briones, T.L.; Darwish, H. Vitamin D mitigates age-related cognitive decline through the modulation of pro-inflammatory state and decrease in amyloid burden. J. Neuroinflamm. 2012, 9, 244. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, K.; Stojanovska, L.; Tangalakis, K.; Bosevski, M.; Apostolopoulos, V. Cognitive decline: A vitamin B perspective. Maturitas 2016, 93, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.W. Vitamin B12 deficiency in the elderly: Is it worth screening? Hong Kong Med. J. 2015, 21, 155–164. [Google Scholar] [CrossRef] [PubMed]
- de Wilde, M.C.; Vellas, B.; Girault, E.; Yavuz, A.C.; Sijben, J.W. Lower brain and blood nutrient status in Alzheimer’s disease: Results from meta-analyses. Alzheimer’s Dement 2017, 3, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.; Roh, H.; Kwon, Y. Causes of hyperhomocysteinemia and its pathological significance. Arch. Pharm. Res. 2018, 41, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Smith, A.D.; Jobst, K.A.; Refsum, H.; Sutton, L.; Ueland, P.M. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch. Neurol. 1998, 55, 1449–1455. [Google Scholar] [CrossRef] [Green Version]
- Van Dam, F.; Van Gool, W.A. Hyperhomocysteinemia and Alzheimer’s disease: A systematic review. Arch. Gerontol. Geriatr. 2009, 48, 425–430. [Google Scholar] [CrossRef]
- Ford, A.H.; Almeida, O.P. Effect of Vitamin B Supplementation on Cognitive Function in the Elderly: A Systematic Review and Meta-Analysis. Drugs Aging 2019, 36, 419–434. [Google Scholar] [CrossRef]
- Zuin, M.; Cervellati, C.; Brombo, G.; Trentini, A.; Roncon, L.; Zuliani, G. Elevated Blood Homocysteine and Risk of Alzheimer’s Dementia: An Updated Systematic Review and Meta-Analysis Based on Prospective Studies. J. Prev. Alzheimers Dis. 2021, 8, 329–334. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schneider, L.S.; Sano, M.; Diaz-Arrastia, R.; van Dyck, C.H.; Weiner, M.F.; Bottiglieri, T.; Jin, S.; Stokes, K.T.; Thomas, R.G.; et al. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 2008, 300, 1774–1783. [Google Scholar] [CrossRef]
- Kwok, T.; Wu, Y.; Lee, J.; Lee, R.; Yung, C.Y.; Choi, G.; Lee, V.; Harrison, J.; Lam, L.; Mok, V. Randomized placebo-controlled trial of using B vitamins to prevent cognitive decline in older mild cognitive impairment patients. Clin. Nutr. 2020, 39, 2399–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.; Zhou, X.; Li, Q.; Zhao, J.; Song, A.; An, P.; Du, Y.; Xu, W.; Huang, G. Effects of Folic Acid and Vitamin B12, Alone and in Combination on Cognitive Function and Inflammatory Factors in the Elderly with Mild Cognitive Impairment: A Single-blind Experimental Design. Curr. Alzheimer Res. 2019, 16, 622–632. [Google Scholar] [CrossRef]
- Chen, H.; Liu, S.; Ge, B.; Zhou, D.; Li, M.; Li, W.; Ma, F.; Liu, Z.; Ji, Y.; Huang, G. Effects of Folic Acid and Vitamin B12 Supplementation on Cognitive Impairment and Inflammation in Patients with Alzheimer’s Disease: A Randomized, Single-Blinded, Placebo-Controlled Trial. J. Prev. Alzheimers Dis. 2021, 8, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Percudani, R.; Peracchi, A. The B6 database: A tool for the description and classification of vitamin B6-dependent enzymatic activities and of the corresponding protein families. BMC Bioinform. 2009, 10, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderón-Ospina, C.A.; Nava-Mesa, M.O. B Vitamins in the nervous system: Current knowledge of the biochemical modes of action and synergies of thiamine, pyridoxine, and cobalamin. CNS Neurosci Ther. 2020, 26, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Asbaghi, O.; Ghanavati, M.; Ashtary-Larky, D.; Bagheri, R.; Rezaei Kelishadi, M.; Nazarian, B.; Nordvall, M.; Wong, A.; Dutheil, F.; Suzuki, K.; et al. Effects of Folic Acid Supplementation on Oxidative Stress Markers: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Antioxidants 2021, 10, 871. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Wu, T.; Zhao, J.; Song, A.; Liu, H.; Xu, W.; Huang, G. Folic acid supplementation improves cognitive function by reducing the levels of peripheral inflammatory cytokines in elderly Chinese subjects with MCI. Sci. Rep. 2016, 6, 37486. [Google Scholar] [CrossRef] [Green Version]
- Gröber, U.; Kisters, K.; Schmidt, J. Neuroenhancement with vitamin B12-underestimated neurological significance. Nutrients 2013, 5, 5031–5045. [Google Scholar] [CrossRef] [Green Version]
- Mosca, P.; Robert, A.; Alberto, J.M.; Meyer, M.; Kundu, U.; Hergalant, S.; Umoret, R.; Coelho, D.; Guéant, J.L.; Leheup, B.; et al. Vitamin B12 Deficiency Dysregulates m6A mRNA Methylation of Genes Involved in Neurological Functions. Mol. Nutr. Food Res. 2021, 65, e2100206. [Google Scholar] [CrossRef]
- Green, R.; Miller, J.W. Vitamin B12 deficiency. Vitam. Horm. 2022, 119, 405–439. [Google Scholar] [CrossRef]
- Theiss, E.L.; Griebsch, L.V.; Lauer, A.A.; Janitschke, D.; Erhardt, V.; Haas, E.C.; Kuppler, K.N.; Radermacher, J.; Walzer, O.; Portius, D.; et al. Vitamin B12 Attenuates Changes in Phospholipid Levels Related to Oxidative Stress in SH-SY5Y Cells. Cells 2022, 11, 2574. [Google Scholar] [CrossRef] [PubMed]
- Politis, A.; Olgiati, P.; Malitas, P.; Albani, D.; Signorini, A.; Polito, L.; De Mauro, S.; Zisaki, A.; Piperi, C.; Stamouli, E.; et al. Vitamin b12 levels in Alzheimer’s disease: Association with clinical features and cytokine production. J. Alzheimers Dis. 2010, 19, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Quan, M.; Li, T.; Jia, J. Serum Homocysteine, Vitamin B12, Folate, and Their Association with Mild Cognitive Impairment and Subtypes of Dementia. J. Alzheimers Dis. 2022, 90, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, L.; Shrestha, B.; Gautam, K.; Khadka, S.; Mahara Rawal, N. Plasma Vitamin B-12 Levels and Risk of Alzheimer’s Disease: A Case-Control Study. Gerontol. Geriatr. Med. 2022, 8, 23337214211057715. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative stress, amyloid-beta peptide, and altered key molecular pathways in the pathogenesis and progression of alzheimer’s disease. J. Alzheim. Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Fernandez, S.; Bobo-Jimenez, V.; Requejo-Aguilar, R.; Gonzalez-Fernandez, S.; Resch, M.; Carabias-Carrasco, M.; Ros, J.; Almeida, A.; Bolanos, J.P. Hippocampal neurons require a large pool of glutathione to sustain dendrite integrity and cognitive function. Redox Biol. 2018, 19, 52–61. [Google Scholar] [CrossRef]
- Wu, L.; Xiong, X.; Wu, X.; Ye, Y.; Jian, Z.; Zhi, Z.; Gu, L. Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front. Mol. Neurosci. 2020, 13, 28. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F. Mitochondria-ros crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 2012, 329635. [Google Scholar] [CrossRef] [Green Version]
- McCleery, J.; Abraham, R.P.; Denton, D.A.; Rutjes, A.W.S.; Chong, L.Y.; Al-Assaf, A.S.; Griffith, D.J.; Rafeeq, S.; Yaman, H.; Malik, M.A.; et al. Vitamin and mineral supplementation for preventing dmentia or delaying cognitive decline in people with mild cognitive impairment. Cochrane Database Syst. Rev. 2018, 11, CD011905. [Google Scholar] [CrossRef]
- Jurcau, A. The Role of Natural Antioxidants in the Prevention of Dementia—Where Do We Stand and Future Perspectives. Nutrients 2021, 13, 282. [Google Scholar] [CrossRef]
- Beydoun, M.A.; Beydoun, H.A.; Fanelli-Kuczmarski, M.T.; Weiss, J.; Hossain, S.; Canas, J.A.; Evans, M.K.; Zonderman, A.B. Association of Serum Antioxidant Vitamins and Carotenoids with Incident Alzheimer Disease and All-Cause Dementia among US Adults. Neurology 2022, 98, e2150–e2162. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, M.A.; Canas, J.A.; Fanelli-Kuczmarski, M.T.; Maldonado, A.I.; Shaked, D.; Kivimaki, M.; Evans, M.K.; Zonderman, A. B Association of antioxidant vitamins A, C, E and carotenoids with cognitive performance over time: A cohort study of middle-aged adults. Nutrients 2020, 12, 3558. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Shi, L.; Zheng, Y.; Sun, Y.; Huang, X.; Zhang, A.; Que, J.; Sun, X.; Shi, J.; Bao, Y.; et al. Leisure Activities and the Risk of Dementia: A Systematic Review and Meta-Analysis. Neurology 2022, 99, e1651–e1663. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.X.; Dong, F.; Wu, L.Y.; Feng, Y.S.; Zhang, F. The beneficial role of exercise on treating Alzheimer’s disease by inhibiting beta-Amyloid peptide. Mol. Neurobiol. 2021, 58, 5890–5906. [Google Scholar] [CrossRef]
- Huuha, A.M.; Norevik, C.S.; Moreira, J.; Kobro-Flatmoen, A.; Scrimgeour, N.; Kivipelto, M.; Van Praag, H.; Ziaei, M.; Sando, S.B.; Wisløff, U.; et al. Can exercise training teach us how to treat Alzheimer’s disease? Ageing Res. Rev. 2022, 75, 101559. [Google Scholar] [CrossRef]
- Jia, R.X.; Liang, J.H.; Xu, Y.; Wang, Y.Q. Effects of physical activity and exercise on the cognitive function of patients with Alzheimer disease: A meta-analysis. BMC Geriatr. 2019, 19, 181. [Google Scholar] [CrossRef]
- Leeuwis, A.E.; Benedictus, M.R.; Kuijer, J.P.A.; Binnewijzend, M.A.A.; Hooghiemstra, A.M.; Verfaillie, S.C.J.; Koene, T.; Scheltens, P.; Barkhof, F.; Prins, N.D.; et al. Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer’s disease. Alzheimer’s Dement. 2017, 13, 531–540. [Google Scholar] [CrossRef]
- De Sousa, R.; Santos, L.G.; Lopes, P.M.; Cavalcante, B.; Improta-Caria, A.C.; Cassilhas, R.C. Physical exercise consequences on memory in obesity: A systematic review. Obes. Rev. 2021, 22, e13298. [Google Scholar] [CrossRef]
- Vidoni, E.D.; Morris, J.K.; Palmer, J.A.; Li, Y.; White, D.; Kueck, P.J.; John, C.S.; Honea, R.A.; Lepping, R.J.; Lee, P.; et al. Dementia risk and dynamic response to exercise: A non-randomized clinical trial. PLoS ONE 2022, 17, e0265860. [Google Scholar] [CrossRef]
- Sofi, F.; Valecchi, D.; Bacci, D.; Abbate, R.; Gensini, G.F.; Casini, A.; Macchi, C. Physical activity and risk of cognitive decline: A meta-analysis of prospective studies. J. Intern. Med. 2011, 269, 107–117. [Google Scholar] [CrossRef]
- Guure, C.B.; Ibrahim, N.A.; Adam, M.B.; Said, S.M. Impact of physical activity on cognitive decline, dementia, and its subtypes: Meta-analysis of prospective studies. Biomed. Res. Int. 2017, 2017, 9016924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Y.; Wang, J.; Cai, X.; Zhang, X.; Zhang, J.; Peng, M.; Xiao, D.; Ouyang, H.; Yan, F. Effects of physical activity interventions on executive function in older adults with dementia: A meta-analysis of randomized controlled trials. Geriatr. Nurs. 2023, 51, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.J.; Yu, A.P.; Bernal, J.; Fong, D.Y.; Chan, D.; Cheng, C.P.; Siu, P.M. Effects of exercise intensity and frequency on improving cognitive performance in middle-aged and older adults with mild cognitive impairment: A pilot randomized controlled trial on the minimum physical activity recommendation from WHO. Front. Physiol. 2022, 13, 1021428. [Google Scholar] [CrossRef]
- Firth, J.; Stubbs, B.; Vancampfort, D.; Schuch, F.; Lagopoulos, J.; Rosenbaum, S.; Ward, P.B. Effect of aerobic exercise on hippocampal volume in humans: A systematic review and meta-analysis. Neuroimage 2018, 166, 230–238. [Google Scholar] [CrossRef]
- Umegaki, H.; Sakurai, T.; Arai, H. Active Life for Brain Health: A Narrative Review of the Mechanism Underlying the Protective Effects of Physical Activity on the Brain. Front. Aging Neurosci. 2021, 13, 761674. [Google Scholar] [CrossRef]
- Rondão, C.; Mota, M.P.; Oliveira, M.M.; Peixoto, F.; Esteves, D. Multicomponent exercise program effects on fitness and cognitive function of elderlies with mild cognitive impairment: Involvement of oxidative stress and BDNF. Front. Aging Neurosci. 2022, 14, 950937. [Google Scholar] [CrossRef]
- Stigger, F.S.; Zago Marcolino, M.A.; Portela, K.M.; Plentz, R. Effects of Exercise on Inflammatory, Oxidative, and Neurotrophic Biomarkers on Cognitively Impaired Individuals Diagnosed with Dementia or Mild Cognitive Impairment: A Systematic Review and Meta-Analysis. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 616–624. [Google Scholar] [CrossRef]
- Enette, L.; Vogel, T.; Merle, S.; Valard-Guiguet, A.G.; Ozier-Lafontaine, N.; Neviere, R.; Leuly-Joncart, C.; Fanon, J.L.; Lang, P.O. Effect of 9 weeks continuous vs. interval aerobic training on plasma BDNF levels, aerobic fitness, cognitive capacity and quality of life among seniors with mild to moderate Alzheimer’s disease: A randomized controlled trial. Eur. Rev. Aging Phys. Act. 2020, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Suzuki, K.; Higuchi, M.; Balogh, L.; Boldogh, I.; Koltai, E. Physical exercise, reactive oxygen species and neuroprotection. Free Radic. Biol. Med. 2016, 98, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, F.; Qin, D.; Chen, H.; Wang, J.; Wang, J.; Song, S.; Wang, C.; Wang, Y.; Liu, S.; et al. The role of brain derived neurotrophic factor in central nervous system. Front. Aging Neurosci. 2022, 14, 986443. [Google Scholar] [CrossRef]
- Mechling, H.; Netz, Y. Aging and inactivity—Capitalizing on the protective effect of planned physical activity in old age. Eur. Rev. Aging Phys. Act. 2009, 6, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, T.; Masurkar, A.V. Gait dysfunction in Alzheimer’s disease. Handb. Clin. Neurol. 2023. [Google Scholar]
- Billot, M.; Calvani, R.; Urtamo, A.; Sánchez-Sánchez, J.L.; Ciccolari-Micaldi, C.; Chang, M.; Roller-Wirnsberger, R.; Wirnsberger, G.; Sinclair, A.; Vaquero-Pinto, N.; et al. Preserving Mobility in Older Adults with Physical Frailty and Sarcopenia: Opportunities, Challenges, and Recommendations for Physical Activity Interventions. Clin. Interv. Aging 2020, 15, 1675–1690. [Google Scholar] [CrossRef]
- Chong, T.; Curran, E.; Southam, J.; Cox, K.L.; Bryant, C.; Goh, A.; You, E.; Ellis, K.A.; Lautenschlager, N.T. Factors Influencing Long-Term Physical Activity Maintenance: A Qualitative Evaluation of a Physical Activity Program for Inactive Older Adults at Risk of Cognitive Decline: The INDIGO Follow-Up Study. J. Alzheimers Dis. 2022, 89, 1025–1037. [Google Scholar] [CrossRef]
- Tawfik, H.M.; Tsatali, M.; Hassanin, H.I. Pilot feasibility study of cognitive training exercises for Egyptian adults: Proof of concept. Int. J. Geriatr. Psychiatry 2021, 37, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kleineidam, L.; Wolfsgruber, S.; Weyrauch, A.S.; Zulka, L.E.; Forstmeier, S.; Roeske, S.; van den Bussche, H.; Kaduszkiewicz, H.; Wiese, B.; Weyerer, S.; et al. Midlife occupational cognitive requirements protect cognitive function in old age by increasing cognitive reserve. Front. Psychol. 2022, 13, 957308. [Google Scholar] [CrossRef]
- Contador, I.; Alzola, P.; Stern, Y.; de la Torre-Luque, A.; Bermejo-Pareja, F.; Fernández-Calvo, B. Is cognitive reserve associated with the prevention of cognitive decline after stroke? A Systematic review and meta-analysis. Ageing Res. Rev. 2023, 84, 101814. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, T.; Voinescu, A.; Petrini, K.; Stanton Fraser, D. Efficacy and Moderators of Virtual Reality for Cognitive Training in People with Dementia and Mild Cognitive Impairment: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2022, 88, 1341–1370. [Google Scholar] [CrossRef] [PubMed]
- Mehrabi, S.; Muñoz, J.E.; Basharat, A.; Li, Y.; Middleton, L.E.; Cao, S.; Barnett-Cowan, M.; Boger, J. Seas the day: Co-designing immersive virtual reality exergames with exercise professionals and people living with dementia. Alzheimer’s Dement. 2021, 17, e051278. [Google Scholar] [CrossRef]
- Stuerz, K.; Hartmann, S.; Holzner, B.; Bichler, C.S.; Niedermeier, M.; Kopp, M.; Guenther, V. Effects of a Two-Step Cognitive and Relaxation Training Program in Care Home Residents with Mild Cognitive Impairment. Int. J. Environ. Res. Public Health 2022, 19, 8316. [Google Scholar] [CrossRef]
- Li, H.; Li, J.; Li, N.; Li, B.; Wang, P.; Zhou, T. Cognitive intervention for persons with mild cognitive impairment: A meta-analysis. Ageing Res. Rev. 2011, 10, 285–296. [Google Scholar] [CrossRef]
- Hohenfeld, C.; Nellessen, N.; Dogan, I.; Kuhn, H.; Müller, C.; Papa, F.; Ketteler, S.; Goebel, R.; Heinecke, A.; Shah, N.J.; et al. Cognitive Improvement and Brain Changes after Real-Time Functional MRI Neurofeedback Training in Healthy Elderly and Prodromal Alzheimer’s Disease. Front. Neurol. 2017, 8, 384. [Google Scholar] [CrossRef] [Green Version]
- Maeng, S.; Hong, J.P.; Kim, W.H.; Kim, H.; Cho, S.E.; Kang, J.M.; Na, K.S.; Oh, S.H.; Park, J.W.; Bae, J.N.; et al. Effects of Virtual Reality-Based Cognitive Training in the Elderly with and without Mild Cognitive Impairment. Psychiatry Investig. 2021, 18, 619–627. [Google Scholar] [CrossRef]
- Gambella, E.; Margaritini, A.; Benadduci, M.; Rossi, L.; D’Ascoli, P.; Riccardi, G.R.; Pasquini, S.; Civerchia, P.; Pelliccioni, G.; Bevilacqua, R.; et al. An integrated intervention of computerized cognitive training and physical exercise in virtual reality for people with Alzheimer’s disease: The jDome study protocol. Front. Neurol. 2022, 13, 964454. [Google Scholar] [CrossRef] [PubMed]
- Hassandra, M.; Galanis, E.; Hatzigeorgiadis, A.; Goudas, M.; Mouzakidis, C.; Karathanasi, E.M.; Petridou, N.; Tsolaki, M.; Zikas, P.; Evangelou, G.; et al. A Virtual Reality App for Physical and Cognitive Training of Older People with Mild Cognitive Impairment: Mixed Methods Feasibility Study. JMIR Serious Games 2021, 9, e24170. [Google Scholar] [CrossRef]
- Tumurbaatar, B.; Fracassi, A.; Scaduto, P.; Guptarak, J.; Woltjer, R.; Jupiter, D.; Taglialatela, G. Preserved autophagy in cognitively intact non-demented individuals with Alzheimer’s neuropathology. Alzheimer’s Dement. 2023. [Google Scholar] [CrossRef] [PubMed]
- Young-Pearse, T.L.; Lee, H.; Hsieh, Y.C.; Chou, V.; Selkoe, D.J. Moving beyond amyloid and tau to capture the biological heterogeneity of Alzheimer’s disease. Trends Neurosci. 2023, 46, 426–444. [Google Scholar] [CrossRef]
- Lemere, C.A.; Lopera, F.; Kosik, K.S.; Lendon, C.L.; Ossa, J.; Saido, T.C.; Yamaguchi, H.; Ruiz, A.; Martinez, A.; Madrigal, L.; et al. The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat. Med. 1996, 2, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Lopera, F. Clinical Features of Early-Onset Alzheimer Disease in a Large Kindred with an E280A Presenilin-1 Mutation. JAMA 1997, 277, 793–799. [Google Scholar] [CrossRef]
- Vélez, J.I.; Lopera, F.; Sepulveda-Falla, D.; Patel, H.R.; Johar, A.S.; Chuah, A.; Tobón, C.; Rivera, D.; Villegas, A.; Cai, Y.; et al. APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer’s disease. Mol. Psychiatry 2016, 21, 916–924. [Google Scholar] [CrossRef] [Green Version]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Lopera, F.; Marino, C.; Chandrahas, A.S.; O’Hare, M.; Villalba-Moreno, N.D.; Aguillon, D.; Baena, A.; Sanchez, J.S.; Vila-Castelar, C.; Ramirez Gomez, L.; et al. Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat. Med. 2023, 29, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qiao, H.; Wang, Z.; Zhao, W.; Chen, T.; Li, B.; Zhu, L.; Chen, S.; Gu, L.; Wu, Y.; et al. Rationally Design and Acoustically Assemble Human Cerebral Cortex-like Microtissues from hiPSC-derived Neural Progenitors and Neurons. Adv. Mater. 2023. [Google Scholar] [CrossRef] [PubMed]
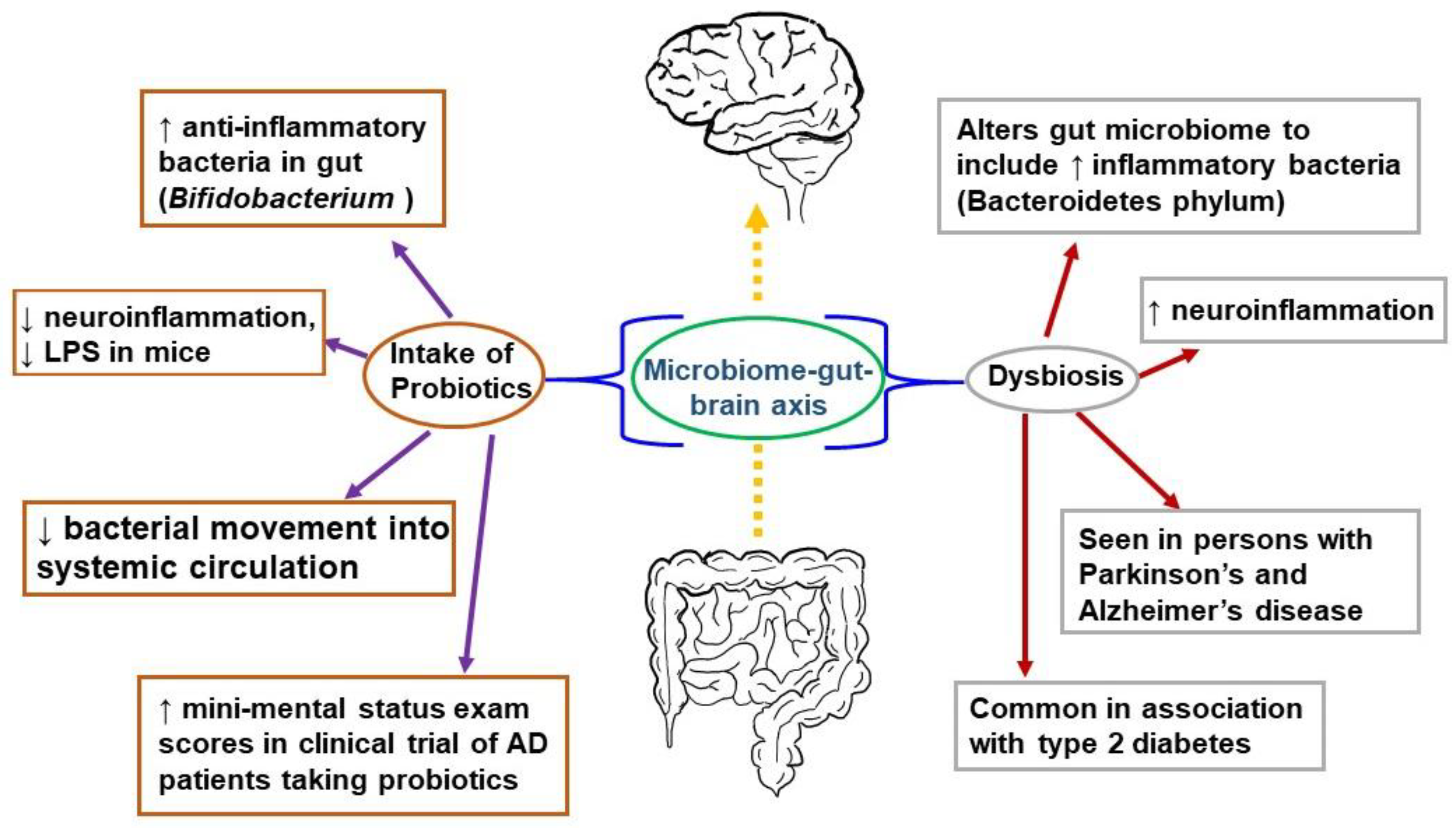
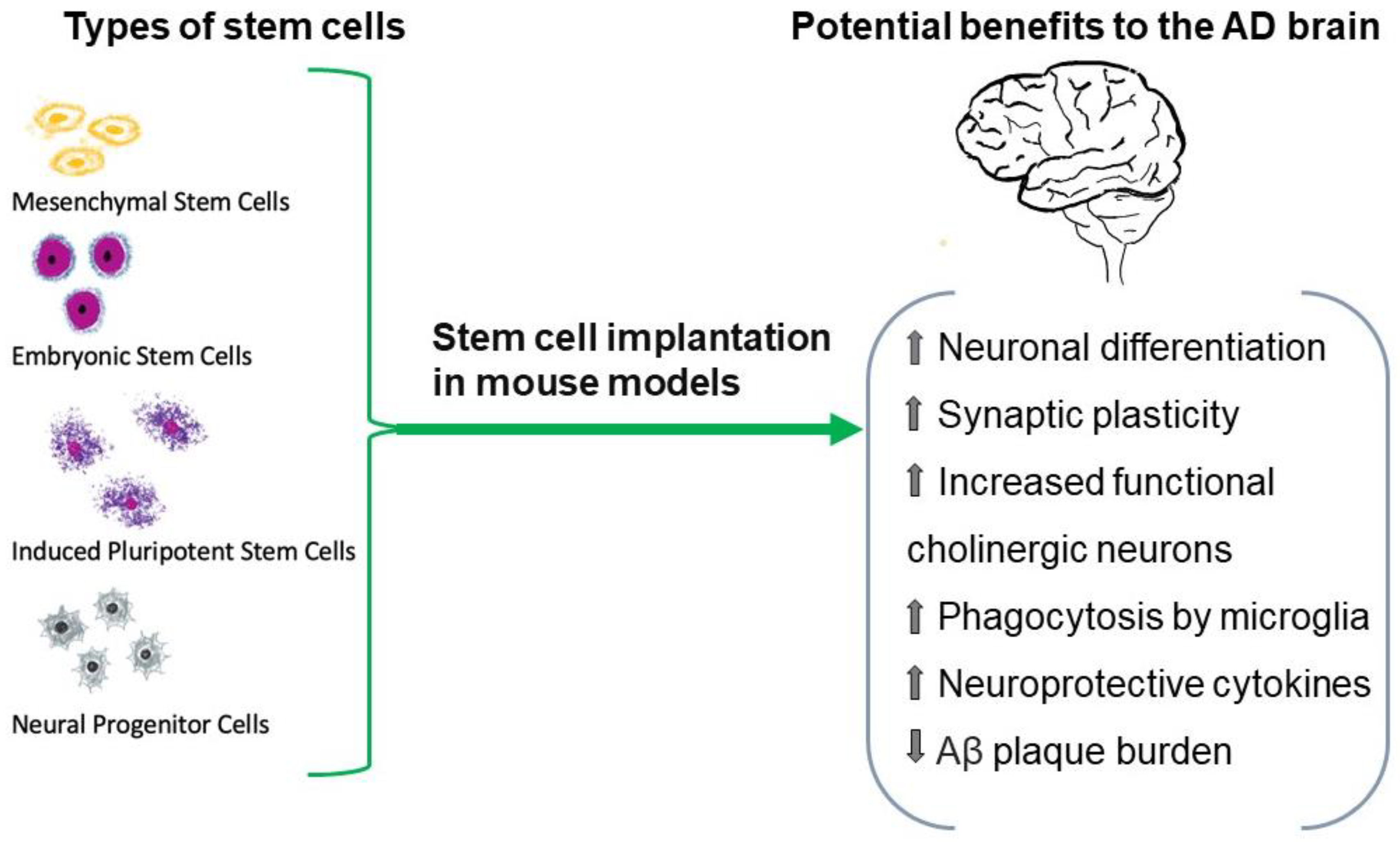

{kind=link}
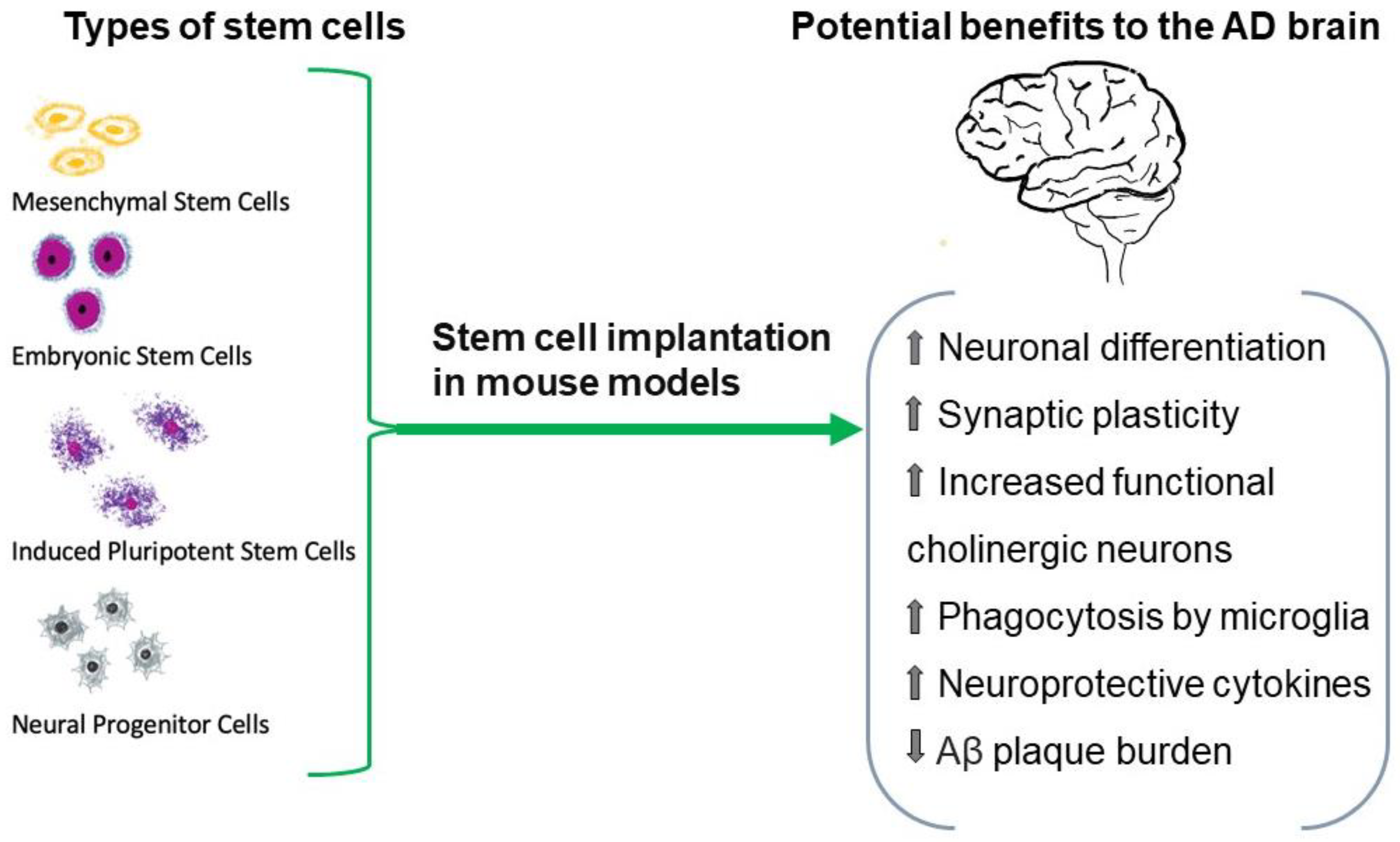{kind=link}
| Targets | Drugs | Modulation of Neuroinflammation |
|---|---|---|
| COX-1 and COX-2 inhibitors | NSAIDs (diclofenac/misoprostol, nimesulide, naproxen, rofecoxib, ibuprofen, indomethacin, tarenflurbil, and celecoxib) | COX-2 overexpression is seen in activated microglia. Potential COX-2 inhibition might reduce neuroinflammatory mediators and prostaglandin release by these cells. |
| TNF-α inhibitors | Etanercept, infliximab, XPro1595 | Activated microglia promote the TNF-α and TNF receptor 1 axis to induce a neuroinflammatory state. |
| TREM2 agonists | (AL002a)—TREM2 mouse IgG1 antibody agonist (AL002c)—mouse IgG1 anti-human TREM2 monoclonal antibody agonist | Genetic mutations in TREM2 receptors are associated with AD. Activation of TREM2 is neuroprotective. |
| CD33 inhibitors | AL003—antibody against CD33 receptor | Higher CD33 levels and subsequent activation of CD33+ microglia are associated with higher Aβ plaque burden. |
| Filamin A conformation restoration | PTI-125—a small molecule drug that interacts with Filamin A to reestablish its native state | Altered filamin A promotes the hyperphosphorylation of tau by activating the signaling of Aβ42 using the α7-nicotinic acetylcholine receptor |
| Category of Method | Specific Intervention | FDA Approved | Clinical Utility or Value | Side Effects | Potentially Disease-Modifying | References |
|---|---|---|---|---|---|---|
| Anti-amyloid | Aducanumab, lecanemab | Accelerated approval | Limited | Infusion reaction, headache, ARIA, brain swelling, brain hemorrhage | Yes | [9,10,11,12,13] |
| Treat CNS insulin resistance | Insulin, metformin | No | Unproven | Hypoglycemia with insulin, GI effects of metformin | Yes | [108,109,110,111,112,113,114,115,116] |
| Stem cells | ESCs, MSCs, iPSCs | No | Unproven | Risks from immunosuppression, tumor formation with ESCs, infection, bleeding | Yes | [161,183,186,187,188,192,193,194] |
| Deep brain stimulation | Delivery of electrical pulses to a defined area of the brain | No | No | Requires implant of the electrode, headache, infection, brain hemorrhage | No | [197,198,199,200,201,202,203,204,205,206,220,221,222] |
| Lifestyle Change | Specific Intervention | Clinical Utility or Value | Side Effects | Potentially Disease-Modifying | References |
|---|---|---|---|---|---|
| Alter gut microbiome | Consumption of probiotics and prebiotics. Fecal transplant. | Unproven | Gas, bloating, constipation, nausea, allergic reactions. | Yes | [130,138,139] |
| Change overall diet | Mediterranean diet, DASH diet, MIND diet | It may preserve memory and lower dementia risk | A Mediterranean diet low in iron | Yes | [230,231,232,233,234,235,236,237,239,240,241] |
| Calorie restriction | Intermittent fasting | Unproven | Hunger, nutritional deficiencies | Maybe | [260,261,262,266,267,268,269] |
| Physical activity, exercise | Structured activity program, non-sedentary lifestyle | May preserve executive function | Risks from falls | Yes | [325,326,327,331,332,333,344] |
| Mental | Cognitive challenges, puzzles, memory tasks, matching tasks, and spatial recognition tasks. | Unproven | None | Maybe | [346,347,348] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiss, A.B.; Muhieddine, D.; Jacob, B.; Mesbah, M.; Pinkhasov, A.; Gomolin, I.H.; Stecker, M.M.; Wisniewski, T.; De Leon, J. Alzheimer’s Disease Treatment: The Search for a Breakthrough. Medicina 2023, 59, 1084. https://doi.org/10.3390/medicina59061084
Reiss AB, Muhieddine D, Jacob B, Mesbah M, Pinkhasov A, Gomolin IH, Stecker MM, Wisniewski T, De Leon J. Alzheimer’s Disease Treatment: The Search for a Breakthrough. Medicina. 2023; 59(6):1084. https://doi.org/10.3390/medicina59061084
Chicago/Turabian StyleReiss, Allison B., Dalia Muhieddine, Berlin Jacob, Michael Mesbah, Aaron Pinkhasov, Irving H. Gomolin, Mark M. Stecker, Thomas Wisniewski, and Joshua De Leon. 2023. "Alzheimer’s Disease Treatment: The Search for a Breakthrough" Medicina 59, no. 6: 1084. https://doi.org/10.3390/medicina59061084
APA StyleReiss, A. B., Muhieddine, D., Jacob, B., Mesbah, M., Pinkhasov, A., Gomolin, I. H., Stecker, M. M., Wisniewski, T., & De Leon, J. (2023). Alzheimer’s Disease Treatment: The Search for a Breakthrough. Medicina, 59(6), 1084. https://doi.org/10.3390/medicina59061084