
Vitamin D Deficiency, Chronic Kidney Disease and Periodontitis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Vitamin D Metabolic Pathways
2.1. Classical Pathway
2.2. Non-Classical Pathway
3. Serum 25(OH)D Thresholds
4. Biological Mechanisms Linking Vitamin D Deficiency, CKD and Periodontitis
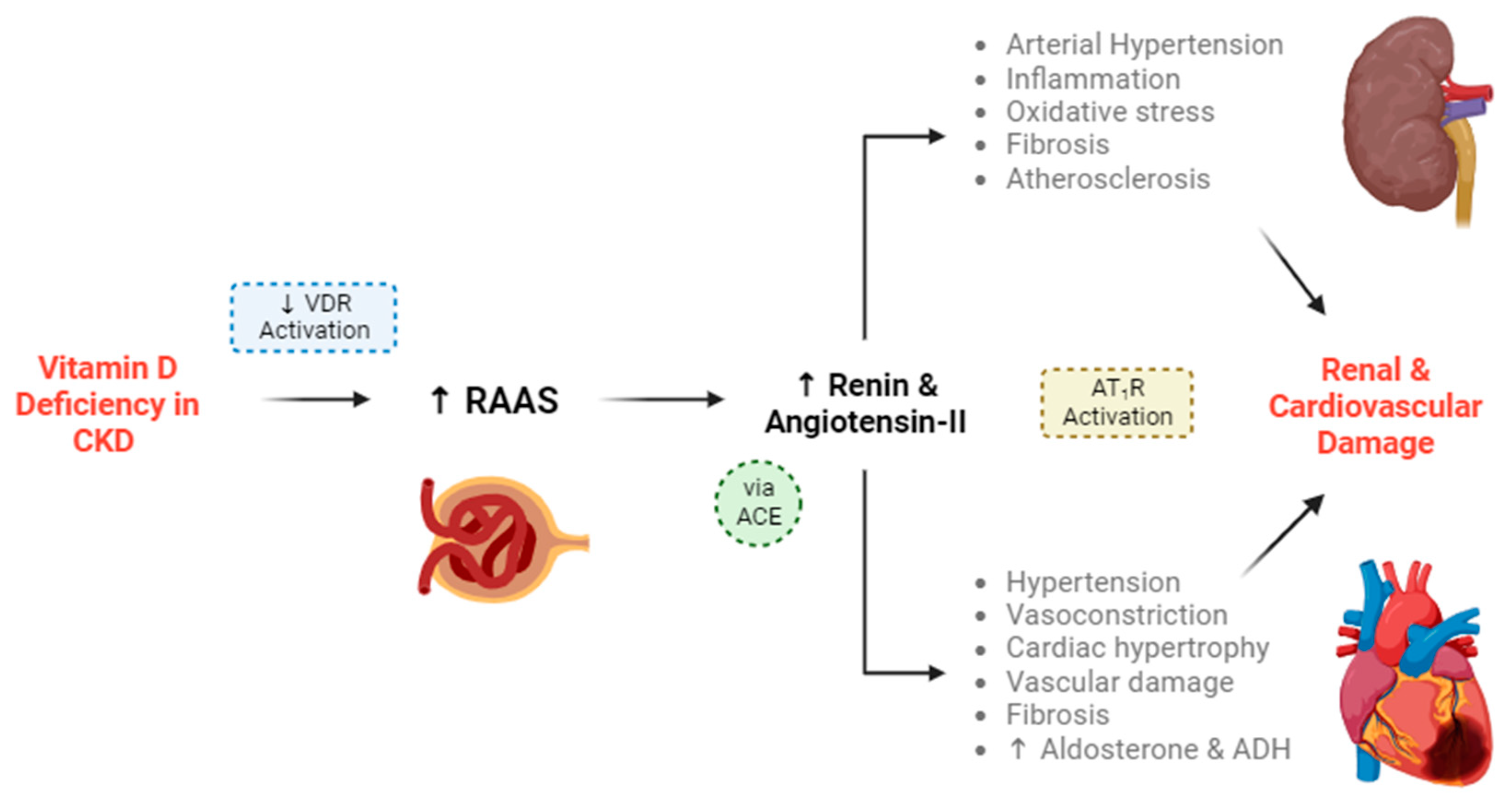
4.1. Altered Vitamin D Pathways in CKD
4.2. Elevations in PTH and FGF23 Levels in CKD
4.3. Oxidative Stress
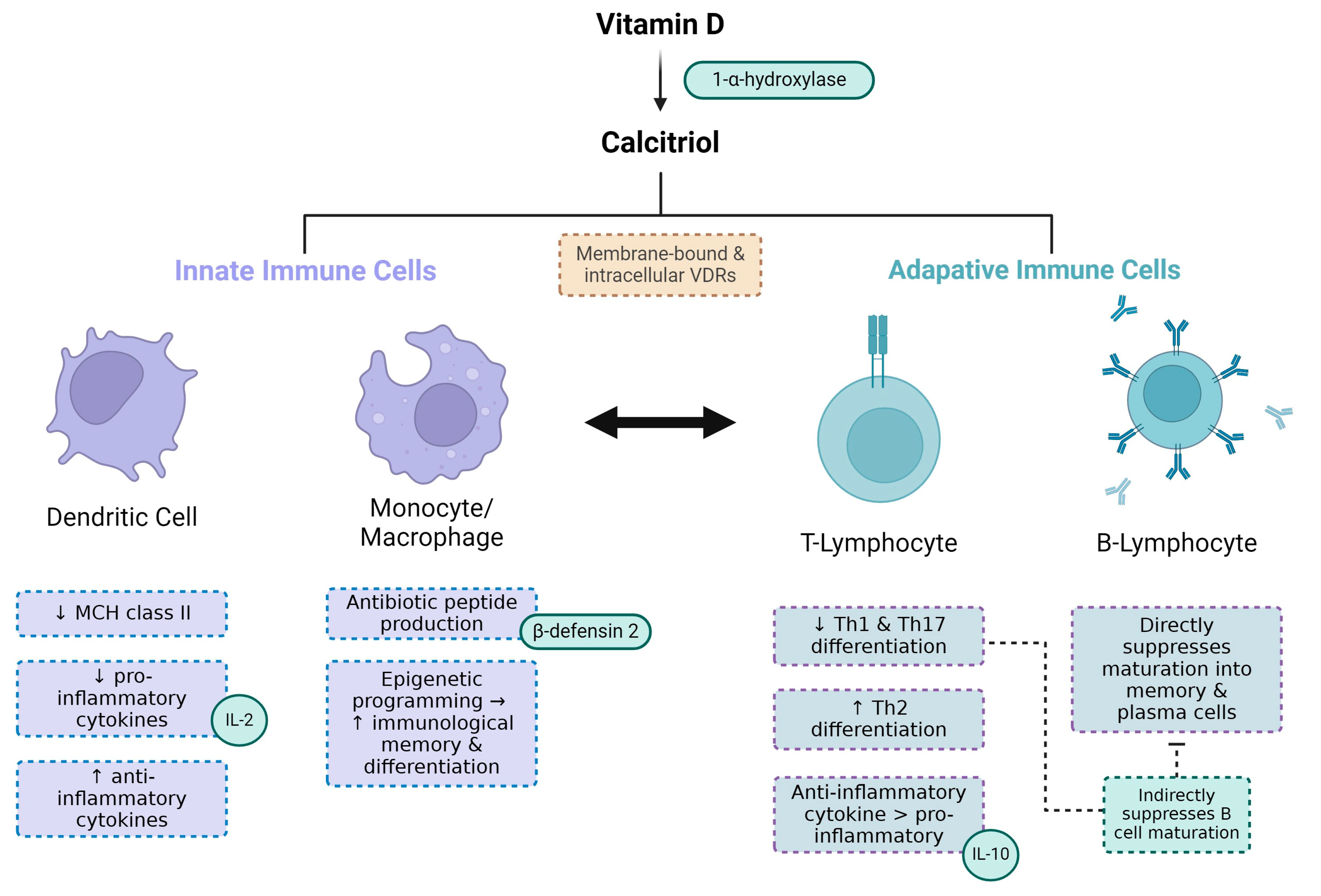
4.4. Impaired Host Response
4.5. DBP Genetic Polymorphisms and Bioavailable 25(OH)D
5. Impact of CKD on Periodontal Inflammation
CKD- Mineral and Bone Disorder and Impact on Periodontitis
6. Impact of Periodontal Inflammation on Renal Function
7. Impact of Vitamin D Deficiency on CKD
Vitamin D Supplementation in CKD Patients
8. Impact of Vitamin D Deficiency on Periodontitis
Impact of Vitamin D Deficiency on Periodontitis in Pregnant Women and Adverse Pregnancy Outcomes
9. Impact of Vitamin D Deficiency on Co-Morbidities Associated with CKD and Periodontitis
9.1. Cardiovascular Disease
9.2. Diabetes Mellitus
9.3. Autoimmune Disease
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lu, E.M.C. The role of vitamin D in periodontal health and disease. J. Periodontal Res. 2023, 58, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Update Work Group. KDIGO 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2017, 7, 1–59. [Google Scholar] [CrossRef] [PubMed]
- Schaeffner, E.S.; Ebert, N.; Delanaye, P.; Frei, U.; Gaedeke, J.; Jakob, O.; Kuhlmann, M.K.; Schuchardt, M.; Tölle, M.; Ziebig, R.; et al. Two novel equations to estimate kidney function in persons aged 70 years or older. Ann. Intern. Med. 2012, 157, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Stevens, P.E.; Levin, A. Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: Synopsis of the kidney disease: Improving global outcomes 2012 clinical practice guideline. Ann. Intern. Med. 2013, 158, 825–830. [Google Scholar] [CrossRef]
- Tonetti, M.S.; Jepsen, S.; Jin, L.; Otomo-Corgel, J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. J. Clin. Periodontol. 2017, 44, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Periodontol. 2018, 89 (Suppl. S1), S173–S182. [Google Scholar] [CrossRef]
- Parsegian, K.; Randall, D.; Curtis, M.; Ioannidou, E. Association between periodontitis and chronic kidney disease. Periodontol. 2000 2022, 89, 114–124. [Google Scholar] [CrossRef]
- Tonetti, M.S.; Van Dyke, T.E.; on behalf of Working Group 1 of the Joint EFP/AAP Workshop. Periodontitis and atherosclerotic cardiovascular disease: Consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J. Periodontol. 2013, 84 (Suppl. S4), S24–S29. [Google Scholar] [CrossRef]
- Deschamps-Lenhardt, S.; Martin-Cabezas, R.; Hannedouche, T.; Huck, O. Association between periodontitis and chronic kidney disease: Systematic review and meta-analysis. Oral Dis. 2019, 25, 385–402. [Google Scholar] [CrossRef]
- Sharma, P.; Fenton, A.; Dias, I.H.K.; Heaton, B.; Brown, C.L.; Sidhu, A.; Rahman, M.; Griffiths, H.R.; Cockwell, P.; Ferro, C.J.; et al. Oxidative stress links periodontal inflammation and renal function. J. Clin. Periodontol. 2021, 48, 357–367. [Google Scholar] [CrossRef]
- Benedik, E. Sources of vitamin D for humans. Int. J. Vitam. Nutr. Res. 2022, 92, 118–125. [Google Scholar] [CrossRef]
- Lehmann, B.; Genehr, T.; Knuschke, P.; Pietzsch, J.; Meurer, M. UVB-Induced Conversion of 7-Dehydrocholesterol to 1α,25-Dihydroxyvitamin D3 in an In Vitro Human Skin Equivalent Model. J. Investig. Dermatol. 2001, 117, 1179–1185. [Google Scholar] [CrossRef]
- Chang, S.W.; Lee, H.C. Vitamin D and health—The missing vitamin in humans. Pediatr. Neonatol. 2019, 60, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Christakos, S.; Ajibade, D.V.; Dhawan, P.; Fechner, A.J.; Mady, L.J. Vitamin D: Metabolism. Endocrinol. Metab. Clin. N. Am. 2010, 39, 243–253. [Google Scholar] [CrossRef]
- Rochel, N.; Wurtz, J.M.; Mitschler, A.; Klaholz, B.; Moras, D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol. Cell. 2000, 5, 173–179. [Google Scholar] [CrossRef]
- Silver, J.; Naveh-Many, T. FGF-23 and secondary hyperparathyroidism in chronic kidney disease. Nat. Rev. Nephrol. 2013, 9, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, A.; Yokoyama, K.; Yokoo, T.; Urashima, M. Akio Nakashima, Keitaro Yokoyama, Takashi Yokoo, Mitsuyoshi Urashima. World J. Diabetes 2016, 7, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Hewison, M. Vitamin D and the immune system: New perspectives on an old theme. Endocrinol. Metab. Clin. N. Am. 2010, 39, 365–379. [Google Scholar] [CrossRef]
- Dusso, A.S. Kidney disease and vitamin D levels: 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, and VDR activation. Kidney Int. Suppl. 2011, 1, 136–141. [Google Scholar] [CrossRef]
- Adams, J.S.; Hewison, M. Update in Vitamin D. J. Clin. Endocrinol. Metab. 2010, 95, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Maranduca, M.A.; Clim, A.; Pinzariu, A.C.; Statescu, C.; Sascau, R.A.; Tanase, D.M.; Serban, D.N.; Branisteanu, D.C.; Branisteanu, D.E.; Huzum, B.; et al. Role of arterial hypertension and angiotensin II in chronic kidney disease (Review). Exp. Ther. Med. 2023, 25, 153. [Google Scholar] [CrossRef] [PubMed]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M. Role of angiotensin II in cardiovascular disease—Therapeutic implications of more than a century of research. JRAAS J. Renin-Angiotensin-Aldosterone Syst. 2006, 7, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.S.; Chen, A.; Ahmad, M.; Wang, H.W.; Leenen, F.H.H. Mineralocorticoid and AT1 receptors in the paraventricular nucleus contribute to sympathetic hyperactivity and cardiac dysfunction in rats post myocardial infarct. J. Physiol. 2014, 592, 3273–3286. [Google Scholar] [CrossRef]
- Iyer, S.N.; Lu, D.; Katovich, M.J.; Raizada, M.K. Chronic control of high blood pressure in the spontaneously hypertensive rat by delivery of angiotensin type 1 receptor antisense. Proc. Natl. Acad. Sci. USA 1996, 93, 9960–9965. [Google Scholar] [CrossRef]
- Wolf, G.; Wenzel, U.; Burns, K.D.; Harris, R.C.; Stahl, R.A.K.; Thaiss, F. Angiotensin II activates nuclear transcription factor-κB through AT1 and AT2 receptors. Kidney Int. 2002, 61, 1986–1995. [Google Scholar] [CrossRef]
- Schieffer, B.; Wirger, A.; Meybrunn, M.; Seitz, S.; Holtz, J.; Riede, U.N.; Drexler, H. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation 1994, 89, 2273–2282. [Google Scholar] [CrossRef]
- Aguilera, G. Role of angiotensin II receptor subtypes on the regulation of aldosterone secretion in the adrenal glomerulosa zone in the rat. Mol. Cell. Endocrinol. 1992, 90, 53–60. [Google Scholar] [CrossRef]
- Qadri, F.; Culman, J.; Veltmar, A.; Maas, K.; Rascher, W.; Unger, T. Angiotensin II-induced vasopressin release is mediated through alpha-1 adrenoceptors and angiotensin II AT1 receptors in the supraoptic nucleus. J. Pharmacol. Exp. Ther. 1993, 267, 567–574. [Google Scholar] [PubMed]
- Sadoshima, J.I.; Izumo, S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts critical role of the AT1 receptor subtype. Circ. Res. 1993, 73, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.; Strömberg, C.; Seltzer, A.; Saavedra, J.M. Balloon angioplasty enhances the expression of angiotensin II AT1 receptors in neointima of rat aorta. J. Clin. Investig. 1992, 90, 1707–1712. [Google Scholar] [CrossRef] [PubMed]
- Jara, Z.P.; Icimoto, M.Y.; Yokota, R.; Ribeiro, A.A.; Dos Santos, F.; Souza, L.E.D.; Watanabe, I.K.M.; Franco, M.D.C.; Pesquero, J.L.; Irigoyen, M.C.; et al. Tonin overexpression in mice diminishes sympathetic autonomic modulation and alters angiotensin type 1 receptor response. Front. Med. 2019, 6, 365. [Google Scholar] [CrossRef]
- Kramár, E.A.; Krishnan, R.; Harding, J.W.; Wright, J.W. Role of nitric oxide in angiotensin IV-induced increases in cerebral blood flow. Regul. Pept. 1998, 74, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.; Malatesta, K.; Norris, K. Vitamin D and Chronic Kidney Disease. Ethn. Dis. 2009, 19, S5–S11. [Google Scholar]
- Freundlich, M.; Quiroz, Y.; Zhang, Z.; Zhang, Y.; Bravo, Y.; Weisinger, J.R.; Li, Y.C.; Rodriguez-Iturbe, B. Suppression of renin–angiotensin gene expression in the kidney by paricalcitol. Kidney Int. 2008, 74, 1394–1402. [Google Scholar] [CrossRef]
- Andress, D.L. Vitamin D in chronic kidney disease: A systemic role for selective vitamin D receptor activation. Kidney Int. 2006, 69, 33–43. [Google Scholar] [CrossRef]
- Tajalli-Nezhad, S.; Karimian, M.; Beyer, C.; Atlasi, M.A.; Tameh, A.A. The regulatory role of Toll-like receptors after ischemic stroke: Neurosteroids as TLR modulators with the focus on TLR2/4. Cell. Mol. Life Sci. 2019, 76, 523–537. [Google Scholar] [CrossRef]
- Wamberg, L.; Kampmann, U.; Stødkilde-Jørgensen, H.; Rejnmark, L.; Pedersen, S.B.; Richelsen, B. Effects of vitamin D supplementation on body fat accumulation, inflammation, and metabolic risk factors in obese adults with low vitamin D levels-results from a randomized trial. Eur. J. Intern. Med. 2013, 24, 644–649. [Google Scholar] [CrossRef]
- Li, Y.C. Renoprotective effects of vitamin D analogs. Kidney Int. 2010, 78, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Hori, M.; Takahashi, H.; Kondo, C.; Hayashi, F.; Tokoroyama, S.; Mori, Y.; Tsujita, M.; Shirasawa, Y.; Takeda, A.; Morozumi, K.; et al. Association between serum 25-hydroxyvitamin D levels and sarcopenia in patients undergoing chronic haemodialysis. Am. J. Nephrol. 2024. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Wintermeyer, E.; Ihle, C.; Ehnert, S.; Stöckle, U.; Ochs, G.; De Zwart, P.; Flesch, I.; Bahrs, C.; Nussler, A.K. Crucial Role of Vitamin D in the Musculoskeletal System. Nutrients 2016, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.U.; Thomas, G.A.; Arnold, A.J. Identification of 1,25-dihydroxyvitamin D3 receptors and activities in muscle. J. Biol. Chem. 1985, 260, 8882–8891. [Google Scholar] [CrossRef] [PubMed]
- Gallieni, M.A.U.R.I.Z.I.O.; Kamimura, S.H.I.G.E.H.I.T.O.; Ahmed, A.D.N.A.N.; Bravo, E.R.I.C.; Delmez, J.A.M.E.S.; Slatopolsky, E.D.U.A.R.D.O.; Dusso, A.D.R.I.A.N.A. Kinetics of monocyte 1 alpha-hydroxylase in renal failure. Am. J. Physiol.-Ren. Physiol. 1995, 268, F746–F753. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, A.; Cuppari, L.; Stenvinkel, P.; Lindholm, B.; Avesani, C.M. Sarcopenia in chronic kidney disease: What have we learned so far? J. Nephrol. 2021, 34, 1347–1372. [Google Scholar] [CrossRef]
- Francis, R.; Aspray, T.; Fraser, W.; Sanjeev Patel, M.; Mavroeidi, A.; Schoenmakers, I.; Stone, M. Vitamin D and Bone Health: A Practical Clinical Guideline for Patient Management. Natl. Osteoporos. Soc. 2018, 2. Available online: https://strwebprdmedia.blob.core.windows.net/media/ef2ideu2/ros-vitamin-d-and-bone-health-in-adults-february-2020.pdf (accessed on 22 January 2024).
- Rosen, C.J.; Abrams, S.A.; Aloia, J.F.; Brannon, P.M.; Clinton, S.K.; Durazo-Arvizu, R.A.; Gallagher, J.C.; Gallo, R.L.; Jones, G.; Kovacs, C.S.; et al. IOM Committee Members Respond to Endocrine Society Vitamin D Guideline. J. Clin. Endocrinol. Metab. 2012, 97, 1146–1152. [Google Scholar] [CrossRef]
- Jones, G. Expanding role for vitamin D in chronic kidney disease: Importance of blood 25-OH-D levels and extra-renal 1alpha-hydroxylase in the classical and nonclassical actions of 1alpha,25-dihydroxyvitamin D(3). Semin. Dial. 2007, 20, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Heaney, R.P. Vitamin D in Health and Disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D status: Measurement, interpretation, and clinical application. Ann. Epidemiol. 2009, 19, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Filipov, J.J.; Zlatkov, B.K.; Dimitrov, E.P.; Svinarov, D. Relationship between vitamin D status and immunosuppressive therapy in kidney transplant recipients. Biotechnol. Biotechnol. Equip. 2015, 29, 331–335. [Google Scholar] [CrossRef]
- Shimada, S.; Hirose, T.; Takahashi, C.; Sato, E.; Kinugasa, S.; Ohsaki, Y.; Kisu, K.; Sato, H.; Ito, S.; Mori, T. Pathophysiological and molecular mechanisms involved in renal congestion in a novel rat model. Sci. Rep. 2018, 8, 16808. [Google Scholar] [CrossRef] [PubMed]
- Faul, C. FGF23 effects on the heart—Levels, time, source, and context matter. Kidney Int. 2018, 94, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef]
- Perwad, F.; Azam, N.; Zhang, M.Y.H.; Yamashita, T.; Tenenhouse, H.S.; Portale, A.A. Dietary and Serum Phosphorus Regulate Fibroblast Growth Factor 23 Expression and 1,25-Dihydroxyvitamin D Metabolism in Mice. Endocrinology. 2005, 146, 5358–5364. [Google Scholar] [CrossRef]
- Usatii, M.; Rousseau, L.; Demers, C.; Petit, J.L.; Brossard, J.H.; Gascon-Barré, M.; Lavigne, J.R.; Zahradnik, R.J.; Nemeth, E.F.; D’amour, P. Parathyroid hormone fragments inhibit active hormone and hypocalcemia-induced 1,25(OH)2D synthesis. Kidney Int. 2007, 72, 1330–1335. [Google Scholar] [CrossRef]
- Szymczak-Pajor, I.; Drzewoski, J.; Śliwińska, A. The Molecular Mechanisms by Which Vitamin D Prevents Insulin Resistance and Associated Disorders. Int. J. Mol. Sci. 2020, 21, 6644. [Google Scholar] [CrossRef]
- Carey, R.M.; Siragy, H.M. Newly Recognized Components of the Renin-Angiotensin System: Potential Roles in Cardiovascular and Renal Regulation. Endocr. Rev. 2003, 24, 261–271. [Google Scholar] [CrossRef]
- Li, P.; He, L.; Sha, Y.; Luan, Q. Relationship of Metabolic Syndrome to Chronic Periodontitis. J. Periodontol. 2009, 80, 541–549. [Google Scholar] [CrossRef]
- Mehrotra, R.; Kermah, D.A.; Salusky, I.B.; Wolf, M.S.; Thadhani, R.I.; Chiu, Y.W.; Martins, D.; Adler, S.G.; Norris, K.C. Chronic kidney disease, hypovitaminosis D, and mortality in the United States. Kidney Int. 2009, 76, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Kestenbaum, B.; Belozeroff, V. Mineral metabolism disturbances in patients with chronic kidney disease. Eur. J. Clin. Investig. 2007, 37, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Ravani, P.; Malberti, F.; Tripepi, G.; Pecchini, P.; Cutrupi, S.; Pizzini, P.; Mallamaci, F.; Zoccali, C. Vitamin D levels and patient outcome in chronic kidney disease. Kidney Int. 2009, 75, 88–95. [Google Scholar] [CrossRef]
- Ho, B.B.; Bergwitz, C. FGF23 signalling and physiology. J. Mol. Endocrinol. 2021, 66, R23–R32. [Google Scholar] [CrossRef] [PubMed]
- Wahl, P.; Wolf, M. FGF23 in chronic kidney disease. Adv. Exp. Med. Biol. 2012, 728, 107–125. [Google Scholar] [CrossRef]
- Jamal, S.A.; Miller, P.D. Secondary and tertiary hyperparathyroidism. J. Clin. Densitom. 2013, 16, 64–68. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Qazi, R.A.; González, E.A.; Zeringue, A.; Martin, K.J. Changes in serum 25-hydroxyvitamin D and plasma intact PTH levels following treatment with ergocalciferol in patients with CKD. Am. J. Kidney Dis. 2007, 50, 59–68. [Google Scholar] [CrossRef]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease—Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef]
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D Activates the Nrf2-Keap1 Antioxidant Pathway and Ameliorates Nephropathy in Diabetic Rats. Am. J. Hypertens. 2014, 27, 586–595. [Google Scholar] [CrossRef]
- Lewis, K.N.; Mele, J.; Hayes, J.D.; Buffenstein, R. Nrf2, a Guardian of Healthspan and Gatekeeper of Species Longevity. Integr. Comp. Biol. 2010, 50, 829–843. [Google Scholar] [CrossRef]
- Tullet, J.M.; Green, J.W.; Au, C.; Benedetto, A.; Thompson, M.A.; Clark, E.; Gilliat, A.F.; Young, A.; Schmeisser, K.; Gems, D. The SKN-1/Nrf2 transcription factor can protect against oxidative stress and increase lifespan in C. elegans by distinct mechanisms. Aging Cell 2017, 16, 1191–1194. [Google Scholar] [CrossRef]
- Razzaque, M.S. FGF23, klotho and vitamin D interactions: What have we learned from in vivo mouse genetics studies? Adv. Exp. Med. Biol. 2012, 728, 84–91. [Google Scholar] [CrossRef]
- Lim, K.; Groen, A.; Molostvov, G.; Lu, T.; Lilley, K.S.; Snead, D.; James, S.; Wilkinson, I.B.; Ting, S.; Hsiao, L.L.; et al. α-Klotho Expression in Human Tissues. J. Clin. Endocrinol. Metab. 2015, 100, E1308–E1318. [Google Scholar] [CrossRef]
- Fan, Y.; Cui, C.; Rosen, C.J.; Sato, T.; Xu, R.; Li, P.; Wei, X.; Bi, R.; Yuan, Q.; Zhou, C. Klotho in Osx+-mesenchymal progenitors exerts pro-osteogenic and anti-inflammatory effects during mandibular alveolar bone formation and repair. Signal Transduct. Target. Ther. 2022, 7, 155. [Google Scholar] [CrossRef]
- Zou, D.; Wu, W.; He, Y.; Ma, S.; Gao, J. The role of klotho in chronic kidney disease. BMC Nephrol. 2018, 19, 285. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.; Bao, D.; Yan, F.; Chen, B. Correlation between serum α-Klotho levels and different stages of periodontitis. BMC Oral Health 2023, 23, 369. [Google Scholar] [CrossRef]
- Zhao, Y.; Banerjee, S.; Dey, N.; LeJeune, W.S.; Sarkar, P.S.; Brobey, R.; Rosenblatt, K.P.; Tilton, R.G.; Choudhary, S. Klotho Depletion Contributes to Increased Inflammation in Kidney of the db/db Mouse Model of Diabetes via RelA (Serine)536 Phosphorylation. Diabetes 2011, 60, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Huang, X.; Fu, C.; Wu, X.; Peng, Y.; Lin, X.; Wang, Y. Recombinant Klotho Protects Human Periodontal Ligament Stem Cells by Regulating Mitochondrial Function and the Antioxidant System during H2O2 -Induced Oxidative Stress. Oxid. Med. Cell. Longev. 2019, 2019, 9261565. [Google Scholar] [CrossRef] [PubMed]
- França, L.F.C.; Vasconcelos, A.C.C.; da Silva, F.R.; Alves, E.H.; Carvalho, J.S.; Lenardo, D.D.; de Souza, L.K.; Barbosa, A.L.; Medeiros, J.V.R.; de Oliveira, J.S.; et al. Periodontitis changes renal structures by oxidative stress and lipid peroxidation. J. Clin. Periodontol. 2017, 44, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Gyurászová, M.; Gurecká, R.; Bábíčková, J.; Tóthová, L. Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers. Oxid. Med. Cell. Longev. 2020, 2020, 5478708. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef]
- Hertiš Petek, T.; Petek, T.; Močnik, M.; Marčun Varda, N. Systemic Inflammation, Oxidative Stress and Cardiovascular Health in Children and Adolescents: A Systematic Review. Antioxidants 2022, 11, 894. [Google Scholar] [CrossRef]
- Annuk, M.; Zilmer, M.; Lind, L.; Linde, T.; Fellström, B. Oxidative Stress and Endothelial Function in Chronic Renal Failure. J. Am. Soc. Nephrol. 2001, 12, 2747–2752. [Google Scholar] [CrossRef]
- Locatelli, F.; Canaud, B.; Eckardt, K.U.; Stenvinkel, P.; Wanner, C.; Zoccali, C. Oxidative stress in end-stage renal disease: An emerging threat to patient outcome. Nephrol. Dial. Transplant. 2003, 18, 1272–1280. [Google Scholar] [CrossRef]
- Berer, A.; Stöckl, J.; Majdic, O.; Wagner, T.; Kollars, M.; Lechner, K.; Geissler, K.; Oehler, L. 1,25-Dihydroxyvitamin D3 inhibits dendritic cell differentiation and maturation in vitro. Exp. Hematol. 2000, 28, 575–583. [Google Scholar] [CrossRef]
- Penna, G.; Adorini, L. 1α,25-Dihydroxyvitamin D3 Inhibits Differentiation, Maturation, Activation, and Survival of Dendritic Cells Leading to Impaired Alloreactive T Cell Activation. J. Immunol. 2000, 164, 2405–2411. [Google Scholar] [CrossRef] [PubMed]
- Ao, T.; Kikuta, J.; Ishii, M. The effects of vitamin D on immune system and inflammatory diseases. Biomolecules 2021, 11, 1624. [Google Scholar] [CrossRef] [PubMed]
- Gombart, A.F.; Borregaard, N.; Koeffler, H.P. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 2005, 19, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Yang, C.S.; Jin, H.S.; Kim, K.K.; Lee, Z.W.; Lee, S.H.; Kim, J.M.; Jo, E.K. Vitamin D3 Induces Autophagy in Human Monocytes/Macrophages via Cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Vitamin D Signaling in the Context of Innate Immunity: Focus on Human Monocytes. Front. Immunol. 2019, 10, 2211. [Google Scholar] [CrossRef]
- Skrobot, A.; Demkow, U.; Wachowska, M. Immunomodulatory Role of Vitamin D: A Review. Adv. Exp. Med. Biol. 2018, 1108, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, U.; Wakita, D.; Ohkuri, T.; Chamoto, K.; Kitamura, H.; Iwakura, Y.; Nishimura, T. 1α,25-Dihydroxyvitamin D3 and all-trans retinoic acid synergistically inhibit the differentiation and expansion of Th17 cells. Immunol. Lett. 2010, 134, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sims, G.P.; Chen, X.X.; Gu, Y.Y.; Chen, S.; Lipsky, P.E. Modulatory Effects of 1,25-Dihydroxyvitamin D3 on Human B Cell Differentiation. J. Immunol. 2007, 179, 1634–1647. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.T.; Nestel, F.P.; Bourdeau, V.; Nagai, Y.; Wang, Q.; Liao, J.; Tavera-Mendoza, L.; Lin, R.; Hanrahan, J.W.; Mader, S.; et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J. Immunol. 2004, 173, 2909–2912. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Beckloff, N.; Ryan, L.K. Host defense peptides in the oral cavity and the lung: Similarities and differences. J. Dent. Res. 2008, 87, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Vitamin D: A Micronutrient Regulating Genes. Curr. Pharm. Des. 2019, 25, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Bhan, I.; Powe, C.E.; Berg, A.H.; Ankers, E.; Wenger, J.B.; Karumanchi, S.A.; Thadhani, R.I. Bioavailable vitamin D is more tightly linked to mineral metabolism than total vitamin D in incident hemodialysis patients. Kidney Int. 2012, 82, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Denburg, M.R.; Bhan, I. Vitamin D-Binding Protein in Health and Chronic Kidney Disease. Semin. Dial. 2015, 28, 636–644. [Google Scholar] [CrossRef]
- Dhaif, Y.G.; Garcia-Sanchez, R.; Albuquerque, R.; Lu, E. The association between Vitamin D binding protein levels and periodontal status: A systematic review. J. Periodontal Res. 2023. ahead of print. [Google Scholar] [CrossRef]
- Powe, C.E.; Evans, M.K.; Wenger, J.; Zonderman, A.B.; Berg, A.H.; Nalls, M.; Tamez, H.; Zhang, D.; Bhan, I.; Karumanchi, S.A.; et al. Vitamin D–Binding Protein and Vitamin D Status of Black Americans and White Americans. N. Engl. J. Med. 2013, 369, 1991–2000. [Google Scholar] [CrossRef]
- Robinson-Cohen, C.; Hoofnagle, A.N.; Ix, J.H.; Sachs, M.C.; Tracy, R.P.; Siscovick, D.S.; Kestenbaum, B.R.; de Boer, I.H. Racial differences in the association of serum 25-hydroxyvitamin D concentration with coronary heart disease events. JAMA 2013, 310, 179–188. [Google Scholar] [CrossRef]
- Parikh, A.; Chase, H.S.; Vernocchi, L.; Stern, L. Vitamin D resistance in chronic kidney disease (CKD). BMC Nephrol. 2014, 15, 47. [Google Scholar] [CrossRef]
- Chun, R.F.; Lauridsen, A.L.; Suon, L.; Zella, L.A.; Pike, J.W.; Modlin, R.L.; Martineau, A.R.; Wilkinson, R.J.; Adams, J.; Hewison, M. Vitamin D-binding protein directs monocyte responses to 25-hydroxy- and 1,25-dihydroxyvitamin D. J. Clin. Endocrinol. Metab. 2010, 95, 3368–3376. [Google Scholar] [CrossRef]
- Taylor, G.W.; Sato, M.; Minagawa, K.; Yoshihara, A.; Iwasaki, M.; Ansai, T. Effect of chronic kidney disease on progression of clinical attachment loss in older adults: A 4-year cohort study. J. Periodontol. 2019, 90, 826–833. [Google Scholar] [CrossRef]
- Serni, L.; Caroti, L.; Barbato, L.; Nieri, M.; Serni, S.; Cirami, C.L.; Cairo, F. Association between chronic kidney disease and periodontitis. A systematic review and metanalysis. Oral Dis. 2023, 29, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Cannata-Andía, J.B.; Martín-Carro, B.; Martín-Vírgala, J.; Rodríguez-Carrio, J.; Bande-Fernández, J.J.; Alonso-Montes, C.; Carrillo-López, N. Chronic Kidney Disease-Mineral and Bone Disorders: Pathogenesis and Management. Calcif. Tissue Int. 2020, 108, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Costacurta, M.; Basilicata, M.; Marrone, G.; Di Lauro, M.; Campolattano, V.; Bollero, P.; Docimo, R.; Di Daniele, N.; Noce, A. The Impact of Chronic Kidney Disease on Nutritional Status and Its Possible Relation with Oral Diseases. Nutrients 2022, 14, 2002. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.R.; Chen, N.X.; Ii, V.H.G.; Moe, S.M. E-Mail Adverse Mandibular Bone Effects Associated with Kidney Disease Are Only Partially Corrected with Bisphosphonate and/or Calcium Treatment. Am. J. Nephrol. 2013, 38, 458–464. [Google Scholar] [CrossRef] [PubMed]
- De Vries, T.J.; Huesa, C. The Osteocyte as a Novel Key Player in Understanding Periodontitis Through its Expression of RANKL and Sclerostin: A Review. Curr. Osteoporos. Rep. 2019, 17, 116–121. [Google Scholar] [CrossRef]
- Liu, M.; Kurimoto, P.; Zhang, J.; Niu, Q.T.; Stolina, M.; Dechow, P.C.; Feng, J.Q.; Hesterman, J.; Silva, M.D.; Ominsky, M.S.; et al. Sclerostin and DKK1 Inhibition Preserves and Augments Alveolar Bone Volume and Architecture in Rats with Alveolar Bone Loss. J. Dent. Res. 2018, 97, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Li, T.J.; Wang, R.; Li, Q.Y.; Li, C.Y.; Jiang, L. Sclerostin regulation: A promising therapy for periodontitis by modulating alveolar bone. Chin. Med. J. 2020, 133, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Asamiya, Y.; Tsuchiya, K.; Nitta, K. Role of sclerostin in the pathogenesis of chronic kidney disease-mineral bone disorder. Ren. Replace. Ther. 2016, 2, 8. [Google Scholar] [CrossRef]
- Fisher, M.A.; Taylor, G.W.; West, B.T.; McCarthy, E.T. Bidirectional relationship between chronic kidney and periodontal disease: A study using structural equation modeling. Kidney Int. 2011, 79, 347–355. [Google Scholar] [CrossRef]
- Lertpimonchai, A.; Rattanasiri, S.; Tamsailom, S.; Champaiboon, C.; Ingsathit, A.; Kitiyakara, C.; Limpianunchai, A.; Attia, J.; Sritara, P.; Thakkinstian, A. Periodontitis as the risk factor of chronic kidney disease: Mediation analysis. J. Clin. Periodontol. 2019, 46, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, V.; Vittinghoff, E.; Taylor, G.; Kritz-Silverstein, D.; Powe, N.; Bibbins-Domingo, K.; Ishani, A.; Cummings, S.R. The association of periodontal disease with kidney function decline: A longitudinal retrospective analysis of the MrOS dental study. Nephrol. Dial. Transplant. 2016, 31, 466–472. [Google Scholar] [CrossRef]
- Kajiwara, K.; Sawa, Y.; Fujita, T.; Tamaoki, S. Immunohistochemical study for the expression of leukocyte adhesion molecules, and FGF23 and ACE2 in P. gingivalis LPS-induced diabetic nephropathy. BMC Nephrol. 2021, 22, 1–12. [Google Scholar] [CrossRef]
- Nakano, C.; Hamano, T.; Fujii, N.; Matsui, I.; Tomida, K.; Mikami, S.; Inoue, K.; Obi, Y.; Okada, N.; Tsubakihara, Y.; et al. Combined use of vitamin D status and FGF23 for risk stratification of renal outcome. Clin. J. Am. Soc. Nephrol. 2012, 7, 810–819. [Google Scholar] [CrossRef]
- Wolf, M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012, 82, 737–747. [Google Scholar] [CrossRef]
- Ersin Kalkan, R.; Öngöz Dede, F.; Gökmenoğlu, C.; Kara, C. Salivary fetuin-A, S100A12, and high-sensitivity C-reactive protein levels in periodontal diseases. Oral Dis. 2018, 24, 1554–1561. [Google Scholar] [CrossRef]
- Jirak, P.; Stechemesser, L.; Moré, E.; Franzen, M.; Topf, A.; Mirna, M.; Paar, V.; Pistulli, R.; Kretzschmar, D.; Wernly, B.; et al. Clinical implications of fetuin-A. Adv. Clin. Chem. 2019, 89, 79–130. [Google Scholar] [CrossRef]
- Furugen, R.; Kawasaki, K.; Kitamura, M.; Maeda, T.; Saito, T.; Hayashida, H. Association of low fetuin-A levels with periodontitis in community-dwelling adults. J. Oral Sci. 2020, 62, 67–69. [Google Scholar] [CrossRef]
- Caglar, K.; Yilmaz, M.I.; Saglam, M.; Cakir, E.; Kilic, S.; Sonmez, A.; Eyileten, T.; Yenicesu, M.; Oguz, Y.; Tasar, M.; et al. Serum Fetuin-A Concentration and Endothelial Dysfunction in Chronic Kidney Disease. Nephron Clin. Pract. 2008, 108, c233–c240. [Google Scholar] [CrossRef]
- Zhou, Z.; Ji, Y.; Ju, H.; Chen, H.; Sun, M. Circulating Fetuin-A and Risk of All-Cause Mortality in Patients with Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Front. Physiol. 2019, 10, 966. [Google Scholar] [CrossRef]
- Bassey, P.E.; Numthavaj, P.; Rattanasiri, S.; Sritara, P.; McEvoy, M.; Ongphiphadhanakul, B.; Thakkinstian, A. Causal association pathways between fetuin-A and kidney function: A mediation analysis. J. Int. Med. Res. 2022, 50, 030006052210828. [Google Scholar] [CrossRef]
- Mahendra, J.; Palathingal, P.; Mahendra, L.; Alzahrani, K.J.; Banjer, H.J.; Alsharif, K.F.; Halawani, I.F.; Muralidharan, J.; Annamalai, P.T.; Verma, S.S.; et al. Impact of Red Complex Bacteria and TNF-α Levels on the Diabetic and Renal Status of Chronic Kidney Disease Patients in the Presence and Absence of Periodontitis. Biology 2022, 11, 451. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, H.; Sun, M.; Chen, J. Association between periodontal disease and mortality in people with CKD: A meta-analysis of cohort studies. BMC Nephrol. 2017, 18, 269. [Google Scholar] [CrossRef]
- Tai, Y.H.; Chen, J.T.; Kuo, H.C.; Chang, W.J.; Wu, M.Y.; Dai, Y.X.; Liu, W.C.; Chen, T.J.; Wu, H.L.; Cherng, Y.G. Periodontal disease and risk of mortality and kidney function decline in advanced chronic kidney disease: A nationwide population-based cohort study. Clin. Oral Investig. 2021, 25, 6259–6268. [Google Scholar] [CrossRef] [PubMed]
- Delbove, T.; Gueyffier, F.; Juillard, L.; Kalbacher, E.; Maucort-Boulch, D.; Nony, P.; Grosgogeat, B.; Gritsch, K. Effect of periodontal treatment on the glomerular filtration rate, reduction of inflammatory markers and mortality in patients with chronic kidney disease: A systematic review. PLoS ONE 2021, 16, e0245619. [Google Scholar] [CrossRef] [PubMed]
- da Silva, T.A.; Abreu, L.G.; Esteves Lima, R.P. A meta-analysis on the effect of periodontal treatment on the glomerular filtration rate of chronic kidney disease individuals: A systematic review and meta-analysis was conducted to assess the impact of the periodontal treatment on the glomerular filtration rate of individuals with chronic kidney disease. Spec. Care Dentist. 2021, 41, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Figueredo, C.M.; Lemos, C.; Bregman, R.; Fischer, R.G. Periodontal treatment in patients with chronic kidney disease: A pilot study. J. Periodontal Res. 2017, 52, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Jean, G.; Terrat, J.C.; Vanel, T.; Hurot, J.M.; Lorriaux, C.; Mayor, B.; Chazot, C. Evidence for persistent vitamin D 1-alpha-hydroxylation in hemodialysis patients: Evolution of serum 1,25-dihydroxycholecalciferol after 6 months of 25-hydroxycholecalciferol treatment. Nephron Clin. Pract. 2008, 110, c58–c65. [Google Scholar] [CrossRef]
- Walker, J.P.; Hiramoto, J.S.; Gasper, W.J.; Auyang, P.; Conte, M.S.; Rapp, J.H.; Lovett, D.H.; Owens, C.D. Vitamin D deficiency is associated with mortality and adverse vascular access outcomes in patients with end-stage renal disease. J. Vasc. Surg. 2014, 60, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.G.; Kerlan, V.; Desailloud, R. Non-classical effects of vitamin D: Non-bone effects of vitamin D. Ann. Endocrinol. 2021, 82, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Melamed, M.L.; Astor, B.; Michos, E.D.; Hostetter, T.H.; Powe, N.R.; Muntner, P. 25-Hydroxyvitamin D levels, race, and the progression of kidney disease. J. Am. Soc. Nephrol. 2009, 20, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Pilz, S.; Tomaschitz, A.; März, W.; Drechsler, C.; Ritz, E.; Zittermann, A.; Cavalier, E.; Pieber, T.R.; Lappe, J.M.; Grant, W.B.; et al. Vitamin D, cardiovascular disease and mortality. Clin. Endocrinol. 2011, 75, 575–584. [Google Scholar] [CrossRef]
- Jayedi, A.; Soltani, S.; Shab-Bidar, S. Vitamin D status and all-cause mortality in patients with chronic kidney disease: A systematic review and dose-response meta-analysis. J. Clin. Endocrinol. Metab. 2017, 102, 2136–2145. [Google Scholar] [CrossRef] [PubMed]
- DeVille, J.; Thorp, M.L.; Tobin, L.; Gray, E.; Johnson, E.S.; Smith, D.H. Effect of ergocalciferol supplementation on serum parathyroid hormone and serum 25-hydroxyvitamin D in chronic kidney disease. Nephrology 2006, 11, 555–559. [Google Scholar] [CrossRef]
- Ikizler, T.A.; Cuppari, L. The 2020 Updated KDOQI Clinical Practice Guidelines for Nutrition in Chronic Kidney Disease. Blood Purif. 2021, 50, 667–671. [Google Scholar] [CrossRef]
- Chowdhury, R.; Kunutsor, S.; Vitezova, A.; Oliver-Williams, C.; Chowdhury, S.; Kiefte-de-Jong, J.C.; Khan, H.; Baena, C.P.; Prabhakaran, D.; Hoshen, M.B.; et al. Vitamin D and risk of cause specific death: Systematic review and meta-analysis of observational cohort and randomised intervention studies. BMJ 2014, 348, g1903. [Google Scholar] [CrossRef]
- Duranton, F.; Rodriguez-Ortiz, M.E.; Duny, Y.; Rodriguez, M.; Daurès, J.P.; Argilés, A. Vitamin D treatment and mortality in chronic kidney disease: A systematic review and meta-analysis. Am. J. Nephrol. 2013, 37, 239–248. [Google Scholar] [CrossRef]
- Cheng, S.; Coyne, D. Vitamin D and outcomes in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2007, 16, 77–82. [Google Scholar] [CrossRef]
- Armas, L.A.G.; Hollis, B.W.; Heaney, R.P. Vitamin D2 Is Much Less Effective than Vitamin D3 in Humans. J. Clin. Endocrinol. Metab. 2004, 89, 5387–5391. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, M.; Aspray, T.J.; Schoenmakers, I. Vitamin D Supplementation for Patients with Chronic Kidney Disease: A Systematic Review and Meta-analyses of Trials Investigating the Response to Supplementation and an Overview of Guidelines. Calcif. Tissue Int. 2021, 109, 157–178. [Google Scholar] [CrossRef]
- Dietrich, T.; Joshipura, K.J.; Dawson-Hughes, B.; Bischoff-Ferrari, H.A. Association between serum concentrations of 25-hydroxyvitamin D3 and periodontal disease in the US population. Am. J. Clin. Nutr. 2004, 80, 108–113. [Google Scholar]
- Krall, E.A.; Wehler, C.; Garcia, R.I.; Harris, S.S.; Dawson-Hughes, B. Calcium and vitamin D supplements reduce tooth loss in the elderly. Am. J. Med. 2001, 111, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Tang, H.; Wang, D.; Zhou, X.; Song, Y.; Wang, Z. Effect of short-term vitamin D supplementation after nonsurgical periodontal treatment: A randomized, double-masked, placebo-controlled clinical trial. J. Periodontal Res. 2020, 55, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.M.; Ravishankar, P.L.; Pramod, V.; Rajula, P.B.; Gayathri, K.; Alam, M.K.; Raj, A.T.; Bhandi, S.; Patil, S. Effect of Supplementation of Vitamin D in Patients with Periodontitis Evaluated before and after Nonsurgical Therapy. Biomed. Res. Int. 2022, 2022, 5869676. [Google Scholar] [CrossRef] [PubMed]
- Hiremath, V.; Rao, C.; Naiak, V.; Prasad, K.V.V. Anti-inflammatory effect of vitamin D on gingivitis: A dose response randomised controlled trial. Indian J. Public Health 2013, 57, 29. [Google Scholar] [CrossRef]
- Perić, M.; Maiter, D.; Cavalier, E.; Lasserre, J.F.; Toma, S. The effects of 6 month Vitamin D supplementation during the non-surgical treatment of periodontitis in Vitamin D–deficient patients: A randomised double-blind placebo-controlled study. Nutrients 2020, 12, 2940. [Google Scholar] [CrossRef]
- Bashutski, J.D.; Eber, R.M.; Kinney, J.S.; Benavides, E.; Maitra, S.; Braun, T.M.; Giannobile, W.V.; McCauley, L.K. Teriparatide and Osseous Regeneration in the Oral Cavity. N. Engl. J. Med. 2010, 363, 2396–2405. [Google Scholar] [CrossRef]
- Bashutski, J.D.; Eber, R.M.; Kinney, J.S.; Benavides, E.; Maitra, S.; Braun, T.M.; Giannobile, W.V.; McCauley, L.K. The Impact of Vitamin D Status on Periodontal Surgery Outcomes. J. Dent. Res. 2011, 90, 1007–1012. [Google Scholar] [CrossRef]
- Machado, V.; Lobo, S.; Proença, L.; Mendes, J.J.; Botelho, J. Vitamin D and Periodontitis: A Systematic Review and Meta-Analysis. Nutrients 2020, 12, 2177. [Google Scholar] [CrossRef]
- Bastos, J.D.A.; Andrade, L.C.F.D.; Ferreira, A.P.; Barroso, E.D.A.; Daibert, P.D.C.; Barreto, P.L.D.S.; Vilela, E.M.; Marcaccini, A.M.; Colugnati, F.A.B.; Bastos, M.G. Serum levels of vitamin D and chronic periodontitis in patients with chronic kidney disease. J. Bras. Nefrol. 2013, 35, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Bostanci, N. Periodontal health and pregnancy outcomes: Time to deliver. Acta Obstet. Gynecol. Scand. 2023, 102, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Ercan, E.; Eratalay, K.; Deren, O.; Gur, D.; Ozyuncu, O.; Altun, B.; Kanlı, C.; Ozdemir, P.; Akıncıbay, H. Evaluation of periodontal pathogens in amniotic fluid and the role of periodontal disease in pre-term birth and low birth weight. Acta Odontol. Scand. 2013, 71, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Raju, K.; Berens, L. Periodontology and pregnancy: An overview of biomedical and epidemiological evidence. Periodontol. 2000 2021, 87, 132–142. [Google Scholar] [CrossRef]
- Bobetsis, Y.A.; Graziani, F.; Gürsoy, M.; Madianos, P.N. Periodontal disease and adverse pregnancy outcomes. Periodontol. 2000 2020, 83, 154–174. [Google Scholar] [CrossRef] [PubMed]
- Hebisch, G.; Grauaug, A.A.; Neumaier-Wagner, P.M.; Stallmach, T.; Huch, A.; Huch, R. The relationship between cervical dilatation, interleukin-6 and interleukin-8 during term labor. Acta Obstet. Gynecol. Scand. 2001, 80, 840–848. [Google Scholar] [CrossRef]
- Ferrillo, M.; Migliario, M.; Roccuzzo, A.; Molinero-Mourelle, P.; Falcicchio, G.; Umano, G.R.; Pezzotti, F.; Foglio Bonda, P.L.; Calafiore, D.; de Sire, A. Periodontal Disease and Vitamin D Deficiency in Pregnant Women: Which Correlation with Preterm and Low-Weight Birth? J. Clin. Med. 2021, 10, 4578. [Google Scholar] [CrossRef]
- Wang, S.; Xin, X.; Luo, W.; Mo, M.; Si, S.; Shao, B.; Shen, Y.; Cheng, H.; Yu, Y. Association of vitamin D and gene variants in the vitamin D metabolic pathway with preterm birth. Nutrition 2021, 89, 111349. [Google Scholar] [CrossRef]
- Lian, R.H.; Qi, P.A.; Yuan, T.; Yan, P.J.; Qiu, W.W.; Wei, Y.; Hu, Y.G.; Yang, K.H.; Yi, B. Systematic review and meta-analysis of vitamin D deficiency in different pregnancy on preterm birth: Deficiency in middle pregnancy might be at risk. Medicine 2021, 100, e26303. [Google Scholar] [CrossRef]
- MacRae, C.; Mercer, S.W.; Guthrie, B.; Henderson, D. Comorbidity in chronic kidney disease: A large cross-sectional study of prevalence in Scottish primary care. Br. J. Gen. Pract. 2021, 71, e243–e249. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, T.; Sharma, P.; Walter, C.; Weston, P.; Beck, J. The epidemiological evidence behind the association between periodontitis and incident atherosclerotic cardiovascular disease. J. Clin. Periodontol. 2013, 40 (Suppl. S14), S70–S84. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Interconnection of periodontal disease and comorbidities: Evidence, mechanisms, and implications. Periodontol. 2000 2022, 89, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Pencina, M.J.; Booth, S.L.; Jacques, P.F.; Ingelsson, E.; Lanier, K.; Benjamin, E.J.; D’Agostino, R.B.; Wolf, M.; Vasan, R.S. Vitamin D deficiency and risk of cardiovascular disease. Circulation 2008, 117, 503–511. [Google Scholar] [CrossRef]
- Zittermann, A.; Trummer, C.; Theiler-schwetz, V.; Lerchbaum, E.; März, W.; Pilz, S. Vitamin d and cardiovascular disease: An updated narrative review. Int. J. Mol. Sci. 2021, 22, 2896. [Google Scholar] [CrossRef]
- Chen, S.; Glenn, D.J.; Ni, W.; Grigsby, C.L.; Olsen, K.; Nishimoto, M.; Law, C.S.; Gardner, D.G. Expression of the vitamin D receptor is increased in the hypertrophic heart. Hypertension 2008, 52, 1106–1112. [Google Scholar] [CrossRef]
- Zittermann, A.; Schulze Schleithoff, S.; Tenderich, G.; Berthold, H.K.; Körfer, R.; Stehle, P. Low vitamin D status: A contributing factor in the pathogenesis of congestive heart failure? J. Am. Coll. Cardiol. 2003, 41, 105–112. [Google Scholar] [CrossRef]
- Somjen, D.; Weisman, Y.; Kohen, F.; Gayer, B.; Limor, R.; Sharon, O.; Jaccard, N.; Knoll, E.; Stern, N. 25-Hydroxyvitamin D3-1α-hydroxylase is expressed in human vascular smooth muscle cells and is upregulated by parathyroid hormone and estrogenic compounds. Circulation 2005, 111, 1666–1671. [Google Scholar] [CrossRef]
- Friedlaender, M.M.; Kornberg, Z.; Wald, H.; Popovtzer, M.M. Renal effect of vitamin D metabolites: Evidence for the essential role of the 25(OH) group. Am. J. Physiol.-Ren. Physiol. 1983, 244, F674–F678. [Google Scholar] [CrossRef] [PubMed]
- Elidrissy, A.T.H.; Munawarah, M.; Alharbi, K.M. Hypocalcemic rachitic cardiomyopathy in infants. J. Saudi Heart Assoc. 2013, 25, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, A.K.; Chhabra, Y.K.; Mahajan, S. Cardiovascular disease in patients with chronic kidney disease: A neglected subgroup. Heart Asia 2016, 8, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.J.; Raggi, P.; Wolf, M.; Gold, A.M.; Chertow, G.M.; Roe, M.T. Targeting Vascular Calcification in Chronic Kidney Disease. JACC Basic Transl. Sci. 2020, 5, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.; Mandrup-Poulsen, T. An immune origin of type 2 diabetes? Diabetologia 2005, 48, 1038–1050. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Bakris, G.L.; Bilous, R.W.; Chiang, J.L.; De Boer, I.H.; Goldstein-Fuchs, J.; Hirsch, I.B.; Kalantar-Zadeh, K.; Narva, A.S.; Navaneethan, S.D.; et al. Diabetic kidney disease: A report from an ADA consensus conference. Diabetes Care 2014, 37, 2864–2883. [Google Scholar] [CrossRef]
- Fernandez-Juarez, G.; Luno, J.; Barrio, V.; de Vinuesa, S.G.; Praga, M.; Goicoechea, M.; Lahera, V.; Casas, L.; Oliva, J. 25 (OH) vitamin D levels and renal disease progression in patients with type 2 diabetic nephropathy and blockade of the renin-angiotensin system. Clin. J. Am. Soc. Nephrol. 2013, 8, 1870–1876. [Google Scholar] [CrossRef]
- Shultis, W.A.; Weil, E.J.; Looker, H.C.; Curtis, J.M.; Shlossman, M.; Genco, R.J.; Knowler, W.C.; Nelson, R.G. Effect of periodontitis on overt nephropathy and end-stage renal disease in type 2 diabetes. Diabetes Care 2007, 30, 306–311. [Google Scholar] [CrossRef]
- Naruishi, K.; Oishi, K.; Inagaki, Y.; Horibe, M.; Bando, M.; Ninomiya, M.; Kawahara, K.; Minakuchi, J.; Kawashima, S.; Shima, K.; et al. Association between periodontal condition and kidney dysfunction in Japanese adults: A cross-sectional study. Clin. Exp. Dent. Res. 2016, 2, 200–207. [Google Scholar] [CrossRef]
- Andrukhov, O.; Andrukhova, O.; Hulan, U.; Tang, Y.; Bantleon, H.P.; Rausch-Fan, X. Both 25-hydroxyvitamin-D3 and 1,25-dihydroxyvitamin- D3 reduces inflammatory response in human periodontal ligament cells. PLoS ONE 2014, 9, e90301. [Google Scholar] [CrossRef]
- Lips, P.; Eekhoff, M.; van Schoor, N.; Oosterwerff, M.; de Jongh, R.; Krul-Poel, Y.; Simsek, S. Vitamin D and type 2 diabetes. J. Steroid Biochem. Mol. Biol. 2017, 173, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Garach, A.; García-Fontana, B.; Muñoz-Torres, M. Vitamin D status, calcium intake and risk of developing type 2 diabetes: An unresolved issue. Nutrients 2019, 11, 642. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D deficiency and diabetes. Biochem. J. 2017, 474, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D, reactive oxygen species and calcium signalling in ageing and disease. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150434. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Micinski, D. Vitamin D upregulates glutamate cysteine ligase and glutathione reductase, and GSH formation, and decreases ROS and MCP-1 and IL-8 secretion in high-glucose exposed U937 monocytes. Biochem. Biophys. Res. Commun. 2013, 437, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, S.; Porri, D.; De Giuseppe, R.; Manuelli, M.; Alessio, F.; Cena, H. The controversial role of Vitamin D as an antioxidant: Results from randomised controlled trials. Nutr. Res. Rev. 2019, 32, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, D.S.; Kang, S. Vitamin D deficiency impairs glucose-stimulated insulin secretion and increases insulin resistance by reducing PPAR-γ expression in nonobese Type 2 diabetic rats. J. Nutr. Biochem. 2016, 27, 257–265. [Google Scholar] [CrossRef]
- Takiishi, T.; Gysemans, C.; Bouillon, R.; Mathieu, C. Vitamin D and diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 419–446. [Google Scholar] [CrossRef]
- Kang, S.; Tsai, L.T.; Zhou, Y.; Evertts, A.; Xu, S.; Griffin, M.J.; Issner, R.; Whitton, H.J.; Garcia, B.A.; Epstein, C.B.; et al. Identification of nuclear hormone receptor pathways causing insulin resistance by transcriptional and epigenomic analysis. Nat. Cell Biol. 2015, 17, 44–56. [Google Scholar] [CrossRef]
- Ong, L.T.C.; Booth, D.R.; Parnell, G.P. Vitamin D and its Effects on DNA Methylation in Development, Aging, and Disease. Mol. Nutr. Food Res. 2020, 64, e2000437. [Google Scholar] [CrossRef]
- Murdaca, G.; Tonacci, A.; Negrini, S.; Greco, M.; Borro, M.; Puppo, F.; Gangemi, S. Emerging role of vitamin D in autoimmune diseases: An update on evidence and therapeutic implications. Autoimmun. Rev. 2019, 18, 102350. [Google Scholar] [CrossRef]
- Mouterde, G.; Gamon, E.; Rincheval, N.; Lukas, C.; Seror, R.; Berenbaum, F.; Dupuy, A.M.; Daien, C.; Daurès, J.P.; Combe, B. Association Between Vitamin D Deficiency and Disease Activity, Disability, and Radiographic Progression in Early Rheumatoid Arthritis: The ESPOIR Cohort. J. Rheumatol. 2020, 47, 1624–1628. [Google Scholar] [CrossRef]
- Rossini, M.; Maddali Bongi, S.; La Montagna, G.; Minisola, G.; Malavolta, N.; Bernini, L.; Cacace, E.; Sinigaglia, L.; Di Munno, O.; Adami, S. Vitamin D deficiency in rheumatoid arthritis: Prevalence, determinants and associations with disease activity and disability. Arthritis Res. Ther. 2010, 12, R216. [Google Scholar] [CrossRef]
- Caraba, A.; Crişan, V.; Romoşan, I.; Mozoş, I.; Murariu, M. Vitamin D status, disease activity, and endothelial dysfunction in early rheumatoid arthritis patients. Dis. Markers 2017, 2017, 5241012. [Google Scholar] [CrossRef]
- Dankers, W.; Davelaar, N.; Asmawidjaja, P.S.; Mus, A.M.; Hazes, J.M.; Colin, E.M.; Lubberts, E. THU0047 1,25(OH)2d3 and dexamethasone additively suppress synovial fibroblast activation by ccr6+ th memory cells and enhance the effect of tnf-alpha blockade. In Proceedings of the Annual European Congress of Rheumatology, EULAR 2018, Amsterdam, The Netherlands, 13–16 June 2018. [Google Scholar] [CrossRef]
- Mok, C.C. Systemic lupus erythematosus: What should family physicians know in 2018? Hong Kong Med. J. 2018, 24, 501–511. [Google Scholar] [CrossRef]
- Schoindre, Y.; Jallouli, M.; Tanguy, M.L.; Ghillani, P.; Galicier, L.; Aumaître, O.; Francès, C.; Le Guern, V.; Lioté, F.; Smail, A.; et al. Lower Vitamin D levels are associated with higher systemic lupus erythematosus activity, but not predictive of disease flare-up. Lupus Sci. Med. 2014, 1, e000027. [Google Scholar] [CrossRef]
- Linker-Israeli, M.; Elstner, E.; Klinenberg, J.R.; Wallace, D.J.; Koeffler, H.P. Vitamin D3 and its synthetic analogs inhibit the spontaneous in vitro immunoglobulin production by SLE-derived PBMC. Clin. Immunol. 2001, 99, 82–93. [Google Scholar] [CrossRef]
- Sellner, J.; Kraus, J.; Awad, A.; Milo, R.; Hemmer, B.; Stüve, O. The increasing incidence and prevalence of female multiple sclerosis-A critical analysis of potential environmental factors. Autoimmun. Rev. 2011, 10, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Hiremath, G.S.; Cettomai, D.; Baynes, M.; Ratchford, J.N.; Newsome, S.; Harrison, D.; Kerr, D.; Greenberg, B.M.; Calabresi, P.A. Vitamin D status and effect of low-dose cholecalciferol and high-dose ergocalciferol supplementation in multiple sclerosis. Mult. Scler. 2009, 15, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Lemire, J.M.; Ince, A.; Takashima, M. 1,25-dihydroxyvitamin d3 attenuates of expression of experimental murine lupus of MRL/1 mice. Autoimmunity 1992, 12, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Mattner, F.; Smiroldo, S.; Galbiati, F.; Muller, M.; Di Lucia, P.; Poliani, P.L.; Martino, G.; Panina-Bordignon, P.; Adorini, L. Inhibition of Th1 development and treatment of chronic-relapsing experimental allergic encephalomyelitis by a non-hypercalcemic analogue of 1,25-dihydroxyvitamin D3. Eur. J. Immunol. 2000, 30, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Fichna, M.; Żurawek, M.; Januszkiewicz-Lewandowska, D.; Gryczynska, M.; Fichna, P.; Sowinski, J.; Nowak, J. Association of the CYP27B1 C(-1260)A polymorphism with autoimmune Addison’s disease. Exp. Clin. Endocrinol. Diabetes 2010, 118, 544–549. [Google Scholar] [CrossRef]
- Yazici, D.; Yavuz, D.; Tarcin, O.; Sancak, S.; Deyneli, O.; Akalin, S. Vitamin D receptor gene ApaI, TaqI, FokI and BsmI polymorphisms in a group of Turkish patients with Hashimoto’s thyroiditis. Minerva Endocrinol. 2013, 38, 195–201. [Google Scholar] [PubMed]
- Zhang, J.; Li, W.; Liu, J.; Wu, W.; Ouyang, H.; Zhang, Q.; Wang, Y.; Liu, L.; Yang, R.; Liu, X.; et al. Polymorphisms in the vitamin D receptor gene and type 1 diabetes mellitus risk: An update by meta-analysis. Mol. Cell. Endocrinol. 2012, 355, 135–142. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganimusa, I.; Chew, E.; Lu, E.M.-C. Vitamin D Deficiency, Chronic Kidney Disease and Periodontitis. Medicina 2024, 60, 420. https://doi.org/10.3390/medicina60030420
Ganimusa I, Chew E, Lu EM-C. Vitamin D Deficiency, Chronic Kidney Disease and Periodontitis. Medicina. 2024; 60(3):420. https://doi.org/10.3390/medicina60030420
Chicago/Turabian StyleGanimusa, Imaan, Emily Chew, and Emily Ming-Chieh Lu. 2024. "Vitamin D Deficiency, Chronic Kidney Disease and Periodontitis" Medicina 60, no. 3: 420. https://doi.org/10.3390/medicina60030420