
PM2.5 Induces Pyroptosis via Activation of the ROS/NF-κB Signaling Pathway in Bronchial Epithelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents
2.2. Cell Culture and PM2.5 Exposure
2.3. Cell Viability Measurement and Lactate Dehydrogenase (LDH) Release Assay
2.4. Cell Death Assay
2.5. Measurement of Intracellular ROS Levels
2.6. Extraction of Nuclear Protein
2.7. Western Blotting
2.8. Immunofluorescence Assay
2.9. Quantification of IL-1β and IL-18 via an Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Statistical Analysis
3. Results
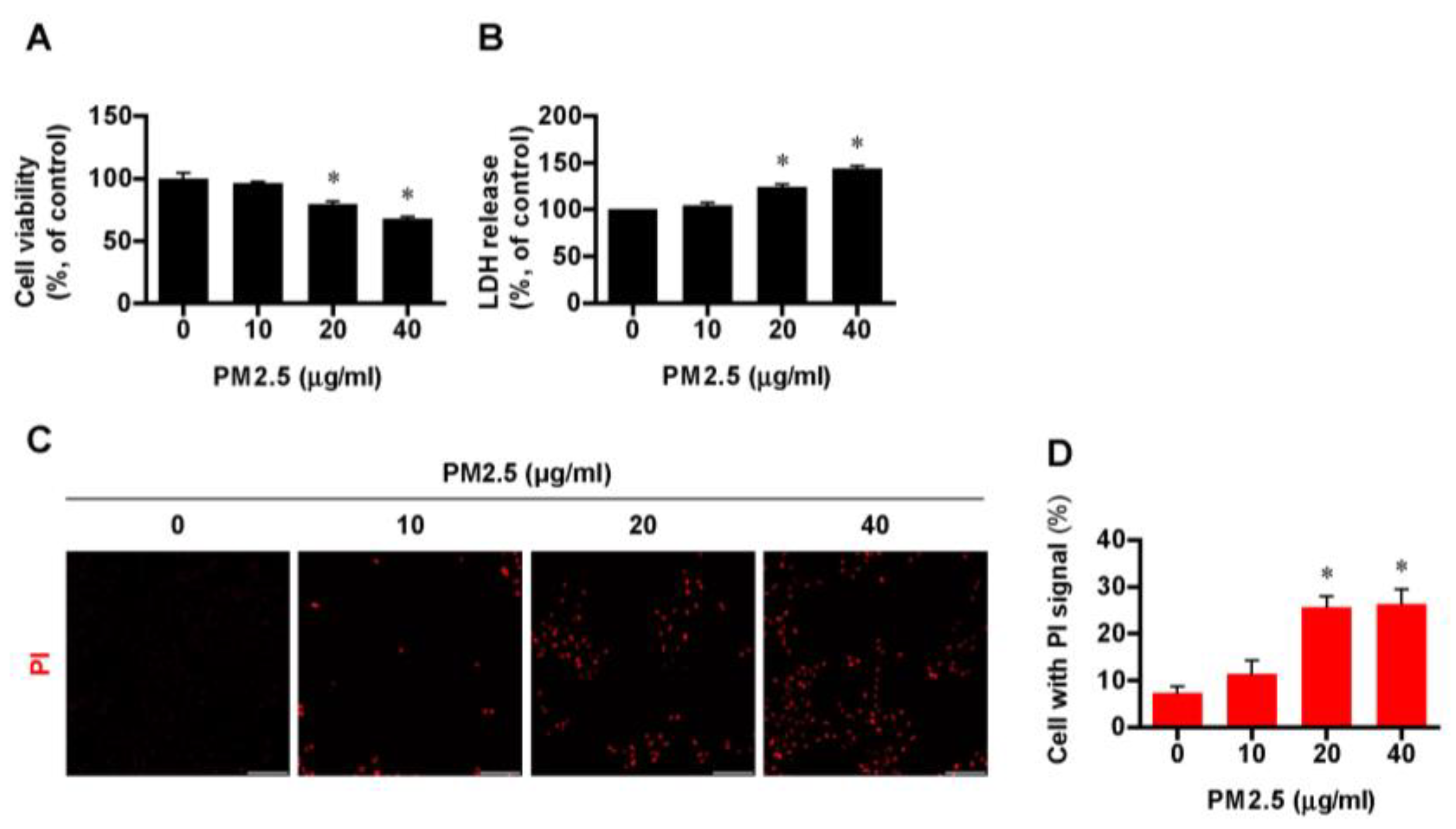
3.1. Exposure to PM2.5 Induced Cell Death in BEAS-2B Cells in a Dose-Dependent Manner
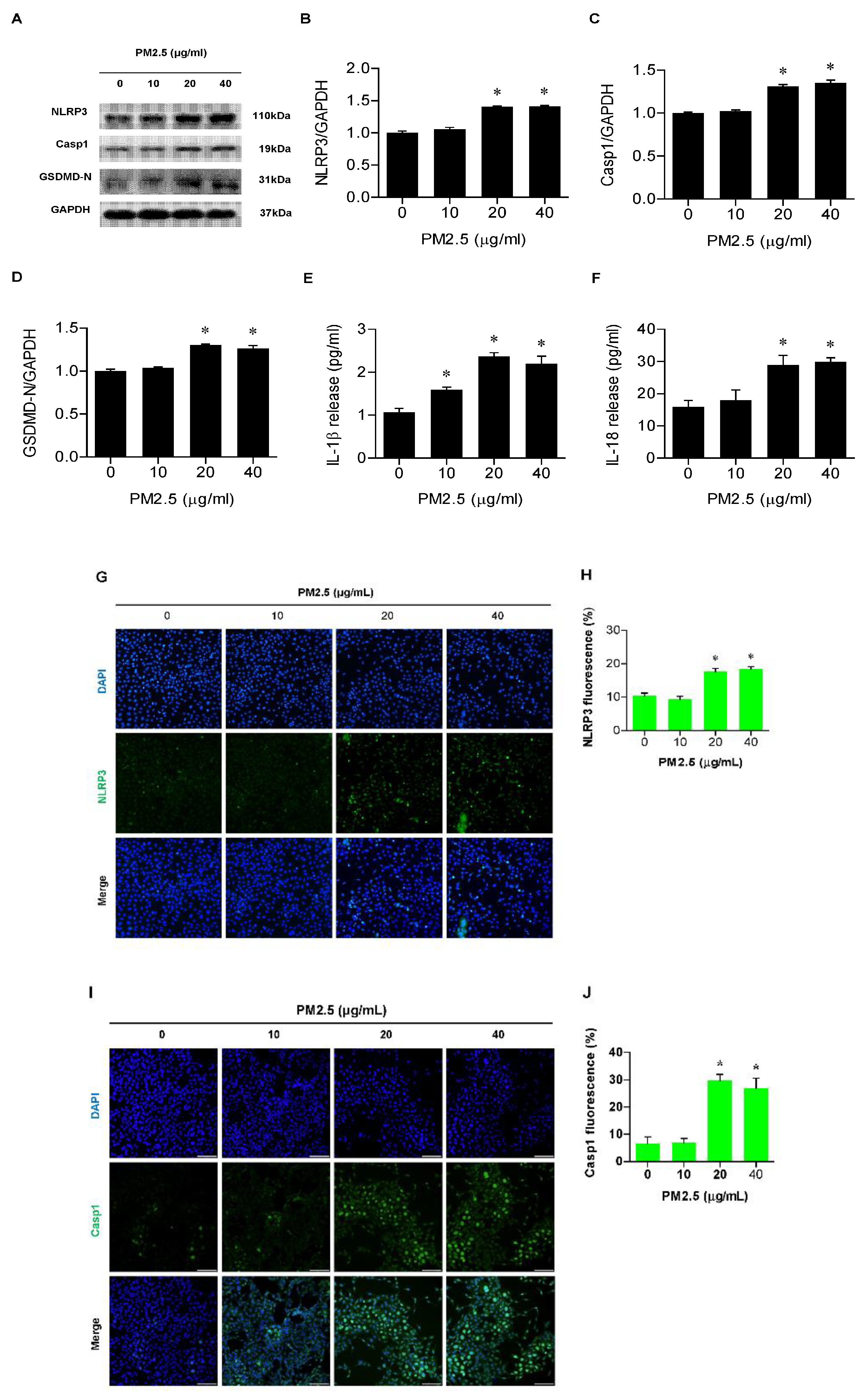
3.2. PM2.5 Involved Activation of the NLRP3 Inflammasome and Release of IL-1β and IL-18 in BEAS-2B Cells
3.3. PM2.5 Exposure Triggered Pyroptosis in BEAS-2B Cells in a NLRP3-Inflammasome-Dependent Manner
3.4. PM2.5 Activated the ROS/NF-ĸB Pathway in BEAS-2B Cells
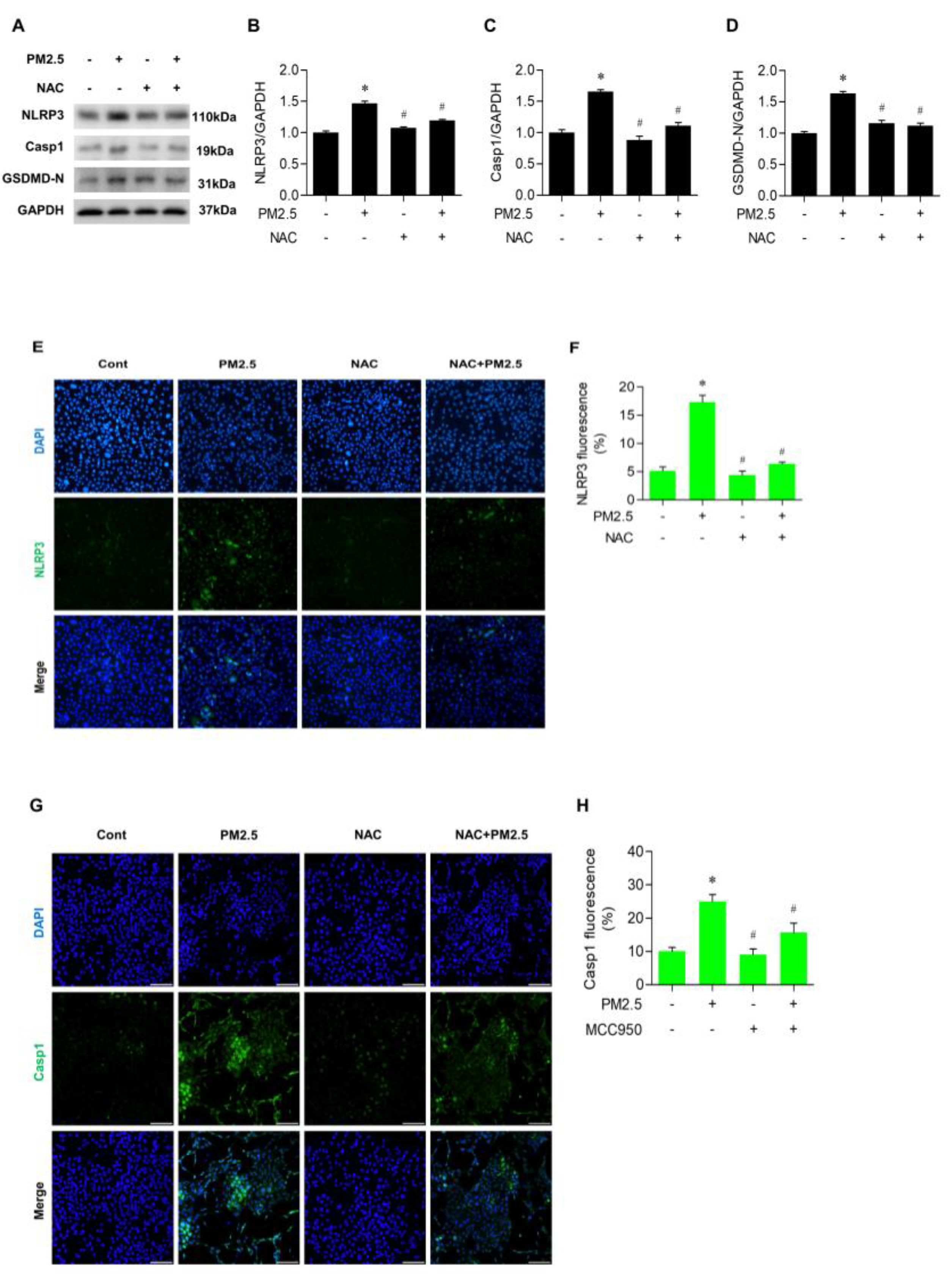
3.5. NAC Inhibited the Expression of NLRP3 and Casp1 in PM2.5-Treated BEAS-2B Cells
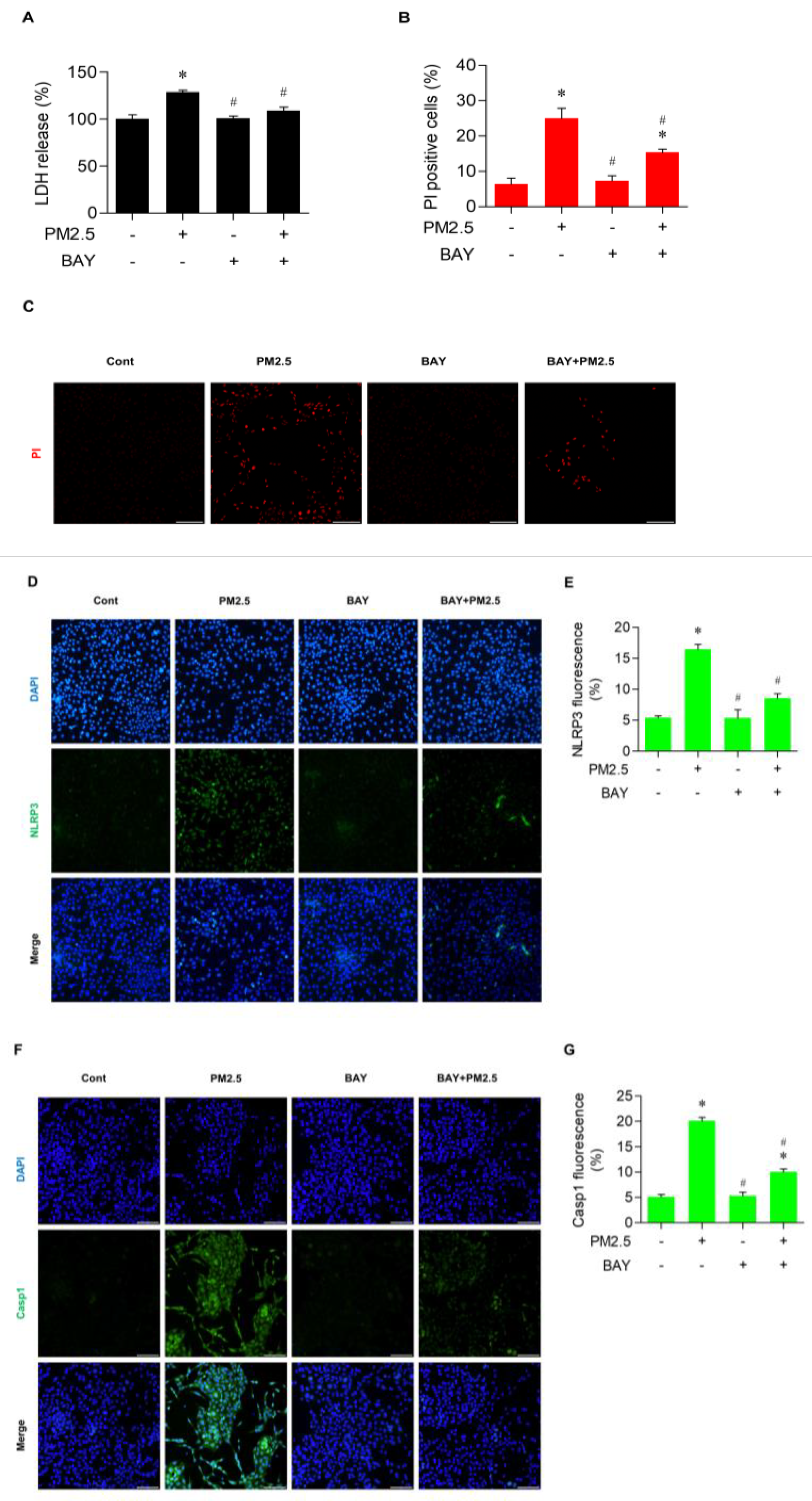
3.6. BAY Diminished Pyroptotic Cell Death in PM2.5-Treated BEAS-2B Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Cui, A.; Xiang, M.; Xu, M.; Lu, P.; Wang, S.; Zou, Y.; Qiao, K.; Jin, C.; Li, Y.; Lu, M.; et al. VCAM-1-mediated neutrophil infiltration exacerbates ambient fine particle-induced lung injury. Toxicol. Lett. 2019, 302, 60–74. [Google Scholar] [CrossRef]
- Health Effects Institute. State of Global Air 2020; Health Effects Institute: Boston, MA, USA, 2020. [Google Scholar]
- Trasande, L.; Malecha, P.; Attina, T.M. Particulate Matter Exposure and Preterm Birth: Estimates of U.S. Attributable Burden and Economic Costs. Environ. Health Perspect. 2016, 124, 1913–1918. [Google Scholar] [CrossRef]
- Cohen, A.J.; Brauer, M.; Burnett, R.; Anderson, H.R.; Frostad, J.; Estep, K.; Balakrishnan, K.; Brunekreef, B.; Dandona, L.; Dandona, R.; et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: An analysis of data from the Global Burden of Diseases Study. Lancet 2017, 389, 1907–1918. [Google Scholar] [CrossRef]
- Li, C.Y.; Wu, C.D.; Pan, W.C.; Chen, Y.C.; Su, H.J. Association Between Long-term Exposure to PM2.5 and Incidence of Type 2 Diabetes in Taiwan: A National Retrospective Cohort Study. Epidemiology 2019, 30 (Suppl. S1), S67–S75. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.C.; Burnett, R.T.; Ezzati, M.; Marshall, J.D.; Robinson, A.L.; Pope, C.A., 3rd. Fine Particulate Matter Exposure and Cancer Incidence: Analysis of SEER Cancer Registry Data from 1992–2016. Environ. Health Perspect. 2020, 128, 107004. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, T.; Zhang, Y.; Chen, H.; Sang, S. Global burden of COPD attributable to ambient PM2.5 in 204 countries and territories, 1990 to 2019: A systematic analysis for the Global Burden of Disease Study. Sci. Total. Environ. 2019, 796, 148819. [Google Scholar] [CrossRef]
- GBD 2019 Risk Factors Collaborators. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study. Lancet 2019, 396, 1223–1249. [Google Scholar] [CrossRef]
- Sang, S.; Chu, C.; Zhang, T.; Chen, H.; Yang, X. The global burden of disease attributable to ambient fine particulate matter in 204 countries and territories, 1990–2019: A systematic analysis of the Global Burden of Disease Study 2019. Ecotoxicol. Environ. Saf. 2022, 238, 113588. [Google Scholar] [CrossRef]
- Risom, L.; Møller, P.; Loft, S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. 2005, 592, 119–137. [Google Scholar] [CrossRef]
- Abbas, I.; Badran, G.; Verdin, A.; Ledoux, F.; Roumie, M.; Lo Guidice, J.M.; Courcot, D.; Garçon, G. In vitro evaluation of organic extractable matter from ambient PM2.5 using human bronchial epithelial BEAS-2B cells: Cytotoxicity, oxidative stress, pro-inflammatory response, genotoxicity, and cell cycle deregulation. Environ. Res. 2019, 171, 510–522. [Google Scholar] [CrossRef]
- Sachdeva, K.; Do, D.C.; Zhang, Y.; Hu, X.; Chen, J.; Gao, P. Environmental Exposures and Asthma Development: Autophagy, Mitophagy, and Cellular Senescence. Front. Immunol. 2019, 10, 2787. [Google Scholar] [CrossRef]
- Kim, J.S.; Choi, H.; Oh, J.M.; Kim, Y.H.; Kim, S.W.; Kim, S.W.; Kim, B.G.; Cho, J.H.; Lee, J.; Lee, D.C. Effect of fluticasone propionate on human nasal fibroblasts exposed to urban particulate matter. Auris Nasus Larynx 2020, 47, 415–424. [Google Scholar] [CrossRef]
- Lee, D.C.; Oh, J.M.; Choi, H.; Kim, S.W.; Kim, S.W.; Kim, B.G.; Cho, J.H.; Lee, J.; Kim, J.-S. Eupatilin Inhibits Reactive Oxygen Species Generation via Akt/NF-κB/MAPK Signaling Pathways in Particulate Matter-Exposed Human Bronchial Epithelial Cells. Toxics 2021, 9, 38. [Google Scholar] [CrossRef]
- Gonzalez-Gonzalez, F.J.; Chandel, N.S.; Jain, M.; Budinger, G.R.S. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl. Res. 2017, 190, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Boukhenouna, S.; Wilson, M.A.; Bahmed, K.; Kosmider, B. Reactive Oxygen Species in Chronic Obstructive Pulmonary Disease. Oxid. Med. Cell. Longev. 2018, 2018, 5730395. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, C.; Tang, X. The Impact of PM2.5 on the Host Defense of Respiratory System. Front. Cell Dev. Biol. 2020, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Choi, H.; Oh, J.M.; Lee, J.; Lee, J.; Lee, H.Y.; Kang, J.Y. Urban particulate matter regulates tight junction proteins by inducing oxidative stress via the Akt signal pathway in human nasal epithelial cells. Toxicol. Lett. 2020, 333, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Feng, Y.; Li, M.; Yangzhong, X.; Zhang, X.; Zu, A.; Hou, Y.; Li, L.; Sun, S. Pyroptosis in inflammation-related respiratory disease. J. Physiol. Biochem. 2022, 78, 721–737. [Google Scholar] [CrossRef]
- Tsai, Y.M.; Chiang, K.H.; Hung, J.Y.; Chang, W.A.; Lin, H.P.; Shieh, J.M.; Chong, I.; Hsu, Y. Der f1 induces pyroptosis in human bronchial epithelia via the NLRP3 inflammasome. Int. J. Mol. Med. 2018, 41, 757–764. [Google Scholar] [CrossRef]
- Liu, T.; Zhou, Y.T.; Wang, L.Q.; Li, L.Y.; Bao, Q.; Tian, S.; Chen, M.X.; Chen, H.X.; Cui, J.; Li, C.W. NOD-like receptor family, pyrin domain containing 3 (NLRP3) contributes to inflammation, pyroptosis, and mucin production in human airway epithelium on rhinovirus infection. J. Allergy Clin. Immunol. 2019, 144, 777–787.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Jiang, Y.X.; Yang, Y.C.; Liu, J.Y.; Huo, C.; Ji, X.L.; Qu, Y.-Q. Cigarette smoke extract induces pyroptosis in human bronchial epithelial cells through the ROS/NLRP3/caspase-1 pathway. Life Sci. 2021, 269, 119090. [Google Scholar] [CrossRef]
- Lee, D.C.; Choi, H.; Oh, J.M.; Hong, Y.; Jeong, S.H.; Kim, C.S.; Kim, D.-K.; Cho, W.-K.; Kim, S.W.; Kim, S.W.; et al. The effect of urban particulate matter on cultured human nasal fibroblasts. Int. Forum Allergy Rhinol. 2018, 8, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Son, K.H.; Heo, H.-R.; Park, E.; Kwak, H.-J.; Uhm, K.-O.; Chung, M.-H.; Kim, Y.-Y.; Lim, H.J. Exogenous 8-Hydroxydeoxyguanosine Attenuates PM2.5-Induced Inflammation in Human Bronchial Epithelial Cells by Decreasing NLRP3 Inflammasome Activation. Antioxidants 2023, 12, 1189. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Y.; Qi, Y.; Liu, S. Cell Death Pathways: The Variable Mechanisms Underlying Fine Particulate Matter-Induced Cytotoxicity. ACS Nanosci. Au 2023, 3, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, J.; Yu, S.; Li, Y.; Zhu, J.; Zhang, K.; Zhang, R. Cell pyroptosis in health and inflammatory diseases. Cell Death Discov. 2022, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; An, Z.; Song, J.; Du, J.; Zhang, L.; Jiang, J.; Ma, Y.; Wang, C.; Zhang, J.; Wu, W. Fine particulate matter-induced lung inflammation is mediated by pyroptosis in mice. Ecotoxicol. Environ. Saf. 2021, 219, 112351. [Google Scholar] [CrossRef]
- Ren, F.; Xu, J.; Zhang, J.; Xu, X.; Huang, L.; Sun, W.; Li, R.; Li, F. PM2.5 induced lung injury through upregulating ROS-dependent NLRP3 Inflammasome-Mediated Pyroptosis. Immunobiology 2022, 227, 152207. [Google Scholar] [CrossRef]
- Hiemstra, P.S.; McCray, P.B., Jr.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef]
- Nam, H.Y.; Choi, B.H.; Lee, J.Y.; Lee, S.G.; Kim, Y.H.; Lee, K.H.; Yoon, H.K.; Song, J.S.; Kim, H.J.; Lim, Y. The role of nitric oxide in the particulate matter (PM2.5)-induced NFkappaB activation in lung epithelial cells. Toxicol. Lett. 2004, 148, 95–102. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Li, D.; Li, X.; Ma, L.; Bai, X.; Wen, Z.; Zhang, X.; Chen, D.; Peng, L. Exposure to PM2.5 induces aberrant activation of NF-κB in human airway epithelial cells by downregulating miR-331 expression. Environ. Toxicol. Pharmacol. 2017, 50, 192–199. [Google Scholar] [CrossRef]
- Dou, C.; Zhang, J.; Qi, C. Cooking oil fume-derived PM2.5 induces apoptosis in A549 cells and MAPK/NF-κB/STAT1 pathway activation. Environ. Sci. Pollut. Res. 2018, 25, 9940–9948. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, U.; Whitsett, J.A.; Wert, S.E.; Tichelaar, J.W.; Bry, K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am. J. Respir. Cell Mol. Biol. 2005, 32, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Ishikawa, Y.; Yoshimoto, T.; Hayashi, N.; Fujimoto, J.; Nakanishi, K. Interleukin 18 acts on memory T helper cells type 1 to induce airway inflammation and hyperresponsiveness in a naive host mouse. J. Exp. Med. 2004, 199, 535–545. [Google Scholar] [CrossRef]
- Moonwiriyakit, A.; Dinsuwannakol, S.; Sontikun, J.; Timpratueang, K.; Muanprasat, C.; Khemawoot, P. Fine particulate matter PM2.5 and its constituent, hexavalent chromium induce acute cytotoxicity in human airway epithelial cells via inflammasome-mediated pyroptosis. Environ. Toxicol. Pharmacol. 2024, 107, 104416. [Google Scholar] [CrossRef]
- Ramos-Martinez, E.; Vega-Sánchez, A.E.; Pérez-Rubio, G.; Mejia, M.; Buendía-Roldán, I.; González-Pérez, M.I.; Mateos-Toledo, H.N.; Andrade, W.A.; Falfán-Valencia, R.; Rojas-Serrano, J. Enhanced Activity of NLRP3 Inflammasome in the Lung of Patients with Anti-Synthetase Syndrome. Cells 2020, 12, 60. [Google Scholar] [CrossRef]
- Guo, Q.; Wu, Y.; Hou, Y.; Liu, Y.; Liu, T.; Zhang, H.; Fan, C.; Guan, H.; Li, Y.; Shan, Z.; et al. Cytokine Secretion and Pyroptosis of Thyroid Follicular Cells Mediated by Enhanced NLRP3, NLRP1, NLRC4, and AIM2 Inflammasomes Are Associated With Autoimmune Thyroiditis. Front. Immunol. 2018, 9, 1197. [Google Scholar] [CrossRef]
- Fusco, R.; Siracusa, R.; Genovese, T.; Cuzzocrea, S.; Di Paola, R. Focus on the Role of NLRP3 Inflammasome in Diseases. Int. J. Mol. Sci. 2020, 21, 4223. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.-Y.; Choi, H.; Oh, J.-M.; Kim, M.; Lee, D.-C. PM2.5 Induces Pyroptosis via Activation of the ROS/NF-κB Signaling Pathway in Bronchial Epithelial Cells. Medicina 2024, 60, 1434. https://doi.org/10.3390/medicina60091434
Kang J-Y, Choi H, Oh J-M, Kim M, Lee D-C. PM2.5 Induces Pyroptosis via Activation of the ROS/NF-κB Signaling Pathway in Bronchial Epithelial Cells. Medicina. 2024; 60(9):1434. https://doi.org/10.3390/medicina60091434
Chicago/Turabian StyleKang, Ji-Young, Hyunsu Choi, Jeong-Min Oh, Minsu Kim, and Dong-Chang Lee. 2024. "PM2.5 Induces Pyroptosis via Activation of the ROS/NF-κB Signaling Pathway in Bronchial Epithelial Cells" Medicina 60, no. 9: 1434. https://doi.org/10.3390/medicina60091434