
Deciphering the Role of Maternal Microchimerism in Offspring Autoimmunity: A Narrative Review

 ,
,  , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Fetal Immune System Development during Pregnancy
3. Self-Tolerance and Autoimmunity
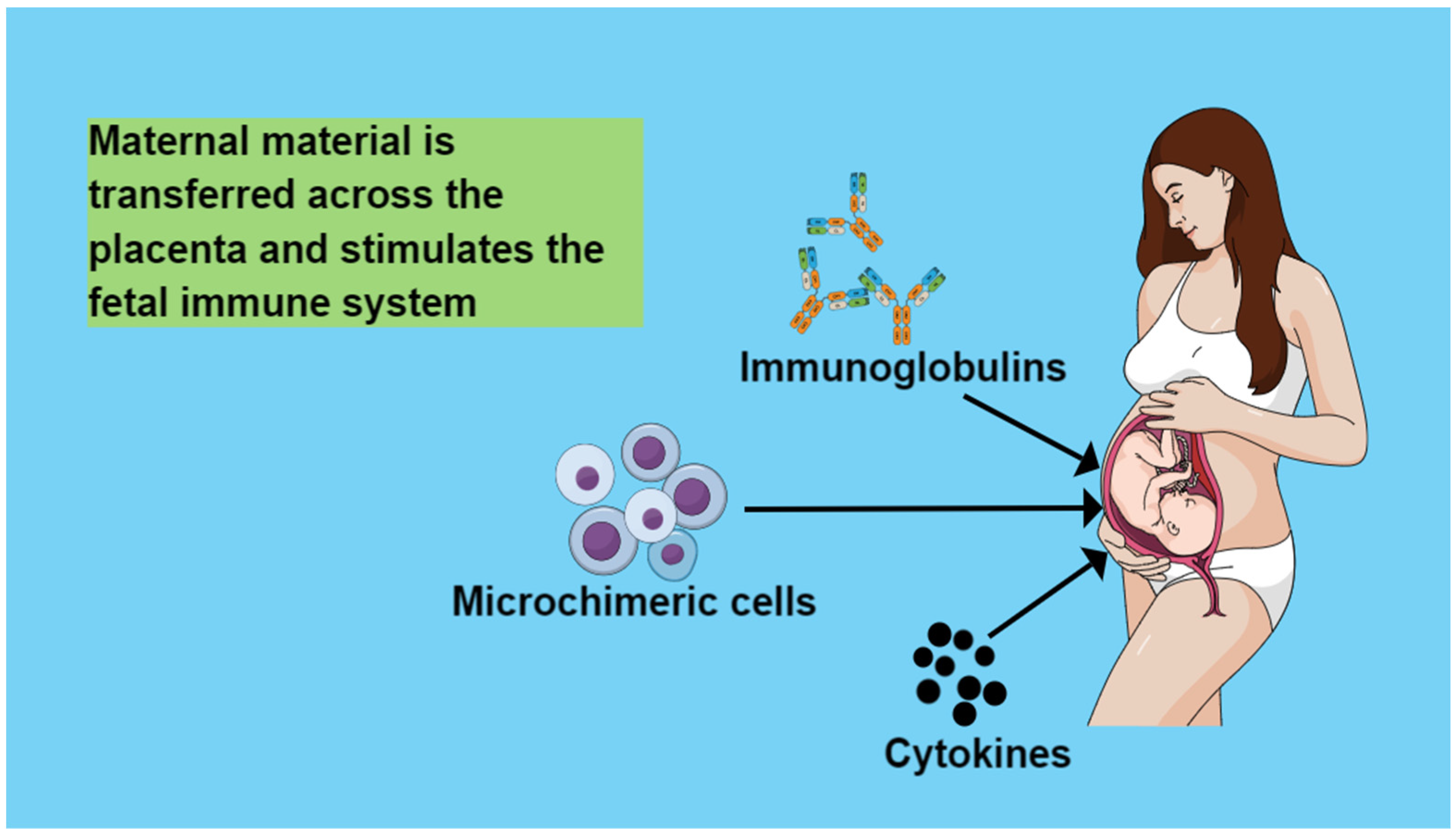
4. Maternal Circulation Material That Is Transferred across the Placenta and Stimulates the Fetal Immune System
4.1. Immunoglobulins
4.2. Cytokines
4.3. Microchimeric Cells
4.3.1. Microchimeric Cell Types
4.3.2. Factors Affecting Prevalence and Incidence of Maternal Microchimeric Cells
4.3.3. Maternal Microchimeric Cells between Generations
4.3.4. The Role of Maternal Microchimerism in Offspring’s Health
4.3.5. Microchimerism and Therapeutic Strategies
4.3.6. Microchimerism and Future Perspectives
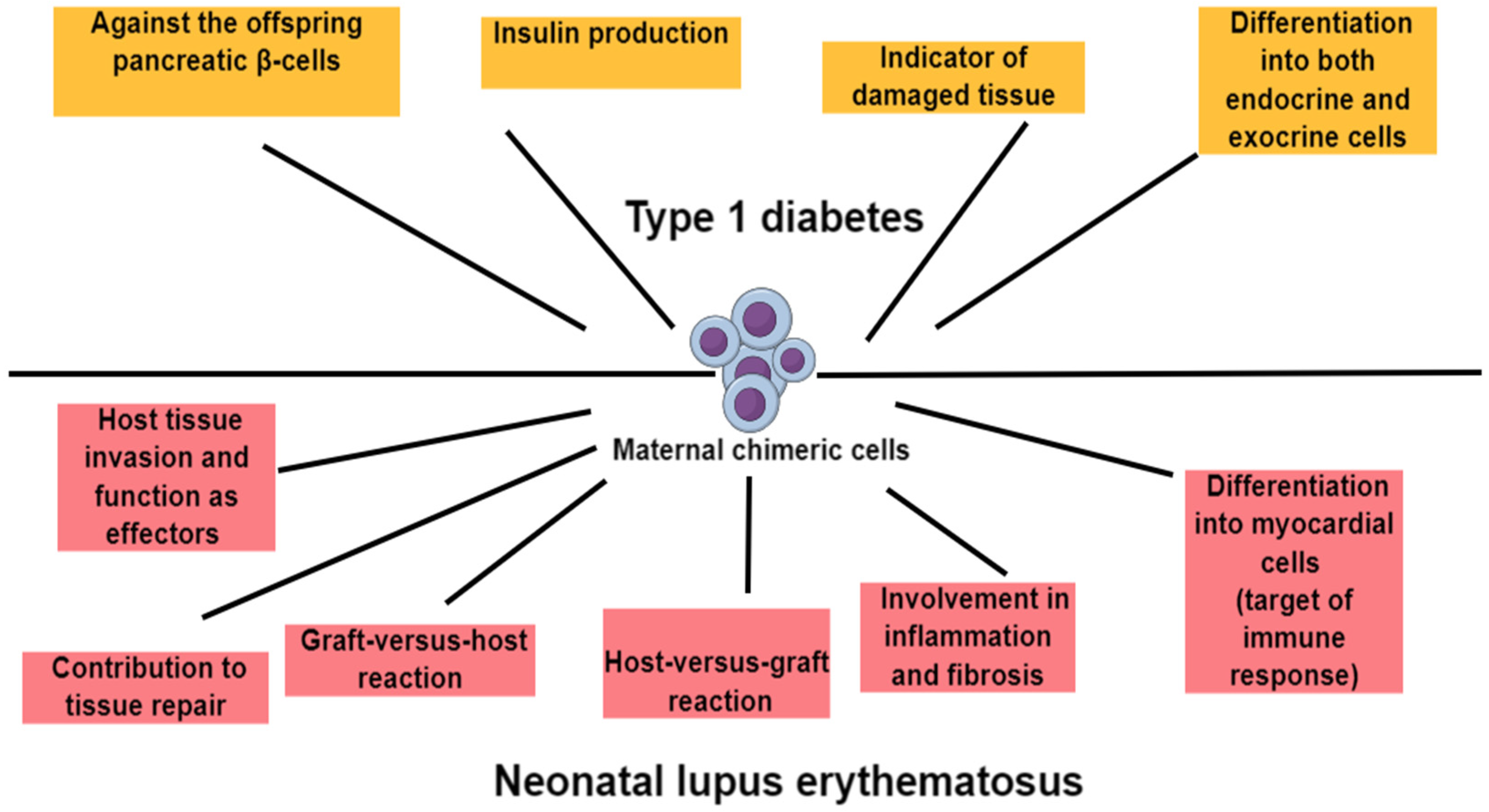
5. Maternal Microchimerism and Offspring’s Autoimmunity
5.1. Maternal Microchimerism and Type 1 Diabetes
5.2. Maternal Microchimerism and Juvenile Idiopathic Inflammatory Myopathies
5.3. Maternal Microchimerism and Systemic Lupus Erythematosus
5.4. Maternal Microchimerism and Sjogren’s Syndrome
6. Future Research
7. Strengths and Limitations
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shrivastava, S.; Naik, R.; Suryawanshi, H.; Gupta, N. Microchimerism: A new concept. J. Oral Maxillofac. Pathol. 2019, 23, 311. [Google Scholar] [CrossRef] [PubMed]
- Madan, K. Natural human chimeras: A review. Eur. J. Med. Genet. 2020, 63, 103971. [Google Scholar] [CrossRef]
- Cómitre-Mariano, B.; Martínez-García, M.; García-Gálvez, B.; Paternina-Die, M.; Desco, M.; Carmona, S.; Gómez-Gaviro, M.V. Feto-maternal microchimerism: Memories from pregnancy. iScience 2021, 25, 103664. [Google Scholar] [CrossRef] [PubMed]
- Gammill, H.S.; Nelson, J.L. Naturally acquired microchimerism. Int. J. Dev. Biol. 2010, 54, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Adams Waldorf, K.M.; Gammill, H.S.; Lucas, J.; Aydelotte, T.M.; Leisenring, W.M.; Lambert, N.C.; Nelson, J.L. Dynamic changes in fetal microchimerism in maternal peripheral blood mononuclear cells, CD4+ and CD8+ cells in normal pregnancy. Placenta 2010, 31, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Kolialexi, A.; Tsangaris, G.T.H.; Antsaklis, A.; Mavrou, A. Rapid clearance of fetal cells from maternal circulation after delivery. Ann. N. Y. Acad. Sci. 2004, 1022, 113–118. [Google Scholar] [CrossRef]
- Mahmood, U.; O’Donoghue, K. Microchimeric fetal cells play a role in maternal wound healing after pregnancy. Chimerism 2014, 5, 40–52. [Google Scholar] [CrossRef]
- Fjeldstad, H.E.S.; Johnsen, G.M.; Staff, A.C. Fetal microchimerism and implications for maternal health. Obstet. Med. 2020, 13, 112–119. [Google Scholar] [CrossRef]
- Jacobsen, D.P.; Fjeldstad, H.E.; Sugulle, M.; Johnsen, G.M.; Olsen, M.B.; Kanaan, S.B.; Staff, A.C. Fetal microchimerism and the two-stage model of preeclampsia. J. Reprod. Immunol. 2023, 159, 104124. [Google Scholar] [CrossRef]
- Boddy, A.M.; Fortunato, A.; Wilson Sayres, M.; Aktipis, A. Fetal microchimerism and maternal health: A review and evolutionary analysis of cooperation and conflict beyond the womb. Bioessays 2015, 37, 1106–1118. [Google Scholar] [CrossRef]
- Kinder, J.M.; Stelzer, I.A.; Arck, P.C.; Way, S.S. Immunological implications of pregnancy-induced microchimerism. Nat. Rev. Immunol. 2017, 17, 483–494. [Google Scholar] [CrossRef]
- Hossain, Z.; Reza, A.H.M.M.; Qasem, W.A.; Friel, J.K.; Omri, A. Development of the immune system in the human embryo. Pediatr. Res. 2022, 92, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.U.; Brodsky, D. Fetal Physiology and the Transition to Extrauterine Life. Clin. Perinatol. 2016, 43, 395–407. [Google Scholar] [CrossRef]
- Migliaccio, G.; Migliaccio, A.R.; Petti, S.; Mavilio, F.; Russo, G.; Lazzaro, D.; Testa, U.; Marinucci, M.; Peschle, C. Human embryonic hemopoiesis. Kinetics of progenitors and precursors underlying the yolk sac----liver transition. J. Clin. Investig. 1986, 78, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.A.; Jen, R.; Brant, R.; Ladd, M.; Huang, Q.; Skoll, A.; Senger, C.; Turvey, S.E.; Marr, N.; Lavoie, P.M. Hierarchical maturation of innate immune defences in very preterm neonates. Neonatology 2014, 106, 1–9. [Google Scholar] [CrossRef]
- Fathman, C.G.; Lineberry, N.B. Molecular mechanisms of CD4+ T-cell anergy. Nat. Rev. Immunol. 2007, 7, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Leveque, L.; Khosrotehrani, K. Can maternal microchimeric cells influence the fetal response toward self antigens? Chimerism 2011, 2, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; Hinterberger, M.; Wirnsberger, G.; Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 2009, 9, 833–844. [Google Scholar] [CrossRef]
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T cells in health and disease. Signal Transduct. Target. Ther. 2023, 8, 235. [Google Scholar] [CrossRef]
- Yang, W.Y.; Shao, Y.; Lopez-Pastrana, J.; Mai, J.; Wang, H.; Yang, X.F. Pathological conditions re-shape physiological Tregs into pathological Tregs. Burn. Trauma 2015, 3, s41038-015-0001-0. [Google Scholar] [CrossRef]
- Kaminitz, A.; Yolcu, E.S.; Stein, J.; Yaniv, I.; Shirwan, H.; Askenasy, N. Killer Treg restore immune homeostasis and suppress autoimmune diabetes in prediabetic NOD mice. J. Autoimmun. 2011, 37, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Hardtke-Wolenski, M.; Landwehr-Kenzel, S. Tipping the balance in autoimmunity: Are regulatory t cells the cause, the cure, or both? Mol. Cell. Pediatr. 2024, 11, 3. [Google Scholar] [CrossRef]
- Chen, J.C. Immunological Consequences of in Utero Exposure to Foreign Antigens. Front. Immunol. 2021, 12, 638435. [Google Scholar] [CrossRef] [PubMed]
- Mold, J.E.; Michaëlsson, J.; Burt, T.D.; Muench, M.O.; Beckerman, K.P.; Busch, M.P.; Lee, T.H.; Nixon, D.F.; McCune, J.M. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science 2008, 322, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Saji, F.; Samejima, Y.; Kamiura, S.; Koyama, M. Dynamics of immunoglobulins at the feto-maternal interface. Rev. Reprod. 1999, 4, 81–89. [Google Scholar] [CrossRef]
- Palmeira, P.; Quinello, C.; Silveira-Lessa, A.L.; Zago, C.A.; Carneiro-Sampaio, M. IgG Placental Transfer in Healthy and Pathological Pregnancies. Clin. Dev. Immunol. 2012, 2012, 985646. [Google Scholar] [CrossRef]
- Simister, N.E.; Story, C.M.; Chen, H.L.; Hunt, J.S. An IgG-transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. Eur. J. Immunol. 1996, 26, 1527–1531. [Google Scholar] [CrossRef]
- Pyzik, M.; Sand, K.M.K.; Hubbard, J.J.; Andersen, J.T.; Sandlie, I.; Blumberg, R.S. The Neonatal Fc Receptor (FcRn): A Misnomer? Front. Immunol. 2019, 10, 1540. [Google Scholar] [CrossRef]
- Malek, A.; Sager, R.; Zakher, A.; Schneider, H. Transport of immunoglobulin G and its subclasses across the in vitro-perfused human placenta. Am. J. Obstet. Gynecol. 1995, 173, 760–767. [Google Scholar] [CrossRef]
- Kim, J.; Hayton, W.L.; Robinson, J.M.; Anderson, C.L. Kinetics of FcRn-mediated recycling of IgG and albumin in human: Pathophysiology and therapeutic implications using a simplified mechanism-based model. Clin. Immunol. 2007, 122, 146–155. [Google Scholar] [CrossRef]
- Bundhoo, A.; Paveglio, S.; Rafti, E.; Dhongade, A.; Blumberg, R.S.; Matson, A.P. Evidence that FcRn mediates the transplacental passage of maternal IgE in the form of IgG anti-IgE/IgE immune complexes. Clin. Exp. Allergy 2015, 45, 1085–1098. [Google Scholar] [CrossRef]
- Brinkhaus, M.; van der Kooi, E.J.; Bentlage, A.E.H.; Ooijevaar-de Heer, P.; Derksen, N.I.L.; Rispens, T.; Vidarsson, G. Human IgE does not bind to human FcRn. Sci. Rep. 2022, 12, 62. [Google Scholar] [CrossRef]
- Lagousi, T.; Gkentzi, D.; Geropeppa, M.; Tsagkli, P.; Spoulou, V. Protecting the Offspring, the Gift of Maternal Immunization: Current Status and Future Perspectives. Vaccines 2022, 10, 1953. [Google Scholar] [CrossRef] [PubMed]
- Gill, T.J.; Repetti, C.F.; Metlay, L.A.; Rabin, B.S.; Taylor, F.H.; Thompson, D.S.; Cortese, A.L. Transplacental immunization of the human fetus to tetanus by immunization of the mother. J. Clin. Investig. 1983, 72, 987–996. [Google Scholar] [CrossRef]
- VANDERBEEKEN, Y.; SARFATI, M.; BOSE, R.; DELESPESSE, G. In utero immunization of the fetus to tetanus by maternal vaccination during pregnancy. Am. J. Reprod. Immunol. Microbiol. 1985, 8, 39–42. [Google Scholar] [CrossRef]
- Rastogi, D.; Wang, C.; Mao, X.; Lendor, C.; Rothman, P.B.; Miller, R.L. Antigen-specific immune responses to influenza vaccine in utero. J. Clin. Investig. 2007, 117, 1637–1646. [Google Scholar] [CrossRef]
- Englund, J.A.; Mbawuike, I.N.; Hammill, H.; Holleman, M.C.; Baxter, B.D.; Glezen, W.P. Maternal immunization with influenza or tetanus toxoid vaccine for passive antibody protection in young infants. J. Infect. Dis. 1993, 168, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.R.; Jones, C.E. Beyond Passive Immunity: Is There Priming of the Fetal Immune System Following Vaccination in Pregnancy and What Are the Potential Clinical Implications? Front. Immunol. 2018, 9, 1548. [Google Scholar] [CrossRef] [PubMed]
- Osman, H.C.; Moreno, R.; Rose, D.; Rowland, M.E.; Ciernia, A.V.; Ashwood, P. Impact of maternal immune activation and sex on placental and fetal brain cytokine and gene expression profiles in a preclinical model of neurodevelopmental disorders. J. Neuroinflamm. 2024, 21, 118. [Google Scholar] [CrossRef]
- Zaretsky, M.V.; Alexander, J.M.; Byrd, W.; Bawdon, R.E. Transfer of inflammatory cytokines across the placenta. Obstet. Gynecol. 2004, 103, 546–550. [Google Scholar] [CrossRef]
- Vidal, M.S.; Menon, R. In utero priming of fetal immune activation: Myths and mechanisms. J. Reprod. Immunol. 2023, 157, 103922. [Google Scholar] [CrossRef]
- Hessami, K.; Tabrizi, R.; Homayoon, N.; Hashemi, A.; Heydari, S.T.; Pourhoseini, S.A. Gestational diabetes mellitus and inflammatory biomarkers of neutrophil-lymphocyte ratio and platelet-lymphocyte ratio: A systematic review and meta-analysis. Biomarkers 2021, 26, 491–498. [Google Scholar] [CrossRef] [PubMed]
- La Verde, M.; Luciano, M.; Fordellone, M.; Sampogna, G.; Lettieri, D.; Palma, M.; Torella, D.; Marrapodi, M.M.; Di Vincenzo, M.; Torella, M. Postpartum Depression and Inflammatory Biomarkers of Neutrophil-Lymphocyte Ratio, Platelet-Lymphocyte Ratio, and Monocyte-Lymphocyte Ratio: A Prospective Observational Study. Gynecol. Obstet. Investig. 2024, 89, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, A.; Cieślik, M.; Adamczyk, A. The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. Int. J. Mol. Sci. 2021, 22, 11516. [Google Scholar] [CrossRef]
- Jonsson, A.M.; Uzunel, M.; Götherström, C.; Papadogiannakis, N.; Westgren, M. Maternal microchimerism in human fetal tissues. Am. J. Obstet. Gynecol. 2008, 198, e1–e325. [Google Scholar] [CrossRef]
- Maloney, S.; Smith, A.; Furst, D.E.; Myerson, D.; Rupert, K.; Evans, P.C.; Nelson, J.L. Microchimerism of maternal origin persists into adult life. J. Clin. Investig. 1999, 104, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, D.W.; Zickwolf, G.K.; Weil, G.J.; Sylvester, S.; Demaria, M.A. Male fetal progenitor cells persist in maternal blood for as long as 27 years postpartum. Proc. Natl. Acad. Sci. USA 1996, 93, 705–708. [Google Scholar] [CrossRef]
- Roy, E.; Leduc, M.; Guegan, S.; Rachdi, L.; Kluger, N.; Scharfmann, R.; Aractingi, S.; Khosrotehrani, K. Specific maternal microchimeric T cells targeting fetal antigens in β cells predispose to auto-immune diabetes in the child. J. Autoimmun. 2011, 36, 253–262. [Google Scholar] [CrossRef]
- Kanold, A.M.J.; Westgren, M.; Götherström, C. Cellular Subsets of Maternal Microchimerism in Umbilical Cord Blood. Cell Transplant. 2019, 28, 522–528. [Google Scholar] [CrossRef]
- Kögler, G.; Göbel, U.; Somville, T.; Enczmann, J.; Arkesteijn, G.; Wernet, P. Simultaneous genotypic and immunophenotypic analysis of interphase cells for the detection of contaminating maternal cells in cord blood and their respective CFU-GM and BFU-E. J. Hematother. 1993, 2, 235–239. [Google Scholar] [CrossRef]
- Kanaan, S.B.; Gammill, H.S.; Harrington, W.E.; De Rosa, S.C.; Stevenson, P.A.; Forsyth, A.M.; Allen, J.; Cousin, E.; van Besien, K.; Delaney, C.S.; et al. Maternal microchimerism is prevalent in cord blood in memory T cells and other cell subsets, and persists post-transplant. Oncoimmunology 2017, 6, e1311436. [Google Scholar] [CrossRef]
- Scaradavou, A.; Carrier, C.; Mollen, N.; Stevens, C.; Rubinstein, P. Detection of Maternal DNA in Placental/Umbilical Cord Blood by Locus-Specific Amplification of the Noninherited Maternal HLA Gene. Blood 1996, 88, 1494–1500. [Google Scholar] [CrossRef]
- Roh, E.Y.; Yoon, J.H.; Shin, S.; Song, E.Y.; Chung, H.Y.; Park, M.H. Frequency of fetal-maternal microchimerism: An analysis of the HLA-DRB1 gene in cord blood and maternal sample pairs. J. Matern. Fetal. Neonatal Med. 2017, 30, 2613–2619. [Google Scholar] [CrossRef] [PubMed]
- El Haddad, M.; Karlmark, K.R.; Donato, X.C.; Martin, G.V.; Bretelle, F.; Lesavre, N.; Cocallemen, J.F.; Martin, M.; Picard, C.; Roudier, J.; et al. Factors Predicting the Presence of Maternal Cells in Cord Blood and Associated Changes in Immune Cell Composition. Front. Immunol. 2021, 12, 651399. [Google Scholar] [CrossRef]
- Berry, S.M.; Hassan, S.S.; Russell, E.; Kukuruga, D.; Land, S.; Kaplan, J. Association of Maternal Histocompatibility at Class II HLA Loci with Maternal Microchimerism in the Fetus. Pediatr. Res. 2004, 56, 73–78. [Google Scholar] [CrossRef]
- Gammill, H.S.; Waldorf, K.M.A.; Aydelotte, T.M.; Lucas, J.; Leisenring, W.M.; Lambert, N.C.; Nelson, J.L. Pregnancy, microchimerism, and the maternal grandmother. PLoS ONE 2011, 6, e24101. [Google Scholar] [CrossRef]
- Haig, D. Does microchimerism mediate kin conflicts? Chimerism 2014, 5, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Schepanski, S.; Chini, M.; Sternemann, V.; Urbschat, C.; Thiele, K.; Sun, T.; Zhao, Y.; Poburski, M.; Woestemeier, A.; Thieme, M.T.; et al. Pregnancy-induced maternal microchimerism shapes neurodevelopment and behavior in mice. Nat. Commun. 2022, 13, 4571. [Google Scholar] [CrossRef]
- Balle, C.; Armistead, B.; Kiravu, A.; Song, X.; Happel, A.U.; Hoffmann, A.A.; Kanaan, S.B.; Nelson, J.L.; Gray, C.M.; Jaspan, H.B.; et al. Factors influencing maternal microchimerism throughout infancy and its impact on infant T cell immunity. J. Clin. Investig. 2022, 132, e148826. [Google Scholar] [CrossRef]
- Castellan, F.S.; Irie, N. Postnatal depletion of maternal cells biases T lymphocytes and natural killer cells’ profiles toward early activation in the spleen. Biol. Open 2022, 11, bio059334. [Google Scholar] [CrossRef]
- Stelzer, I.A.; Urbschat, C.; Schepanski, S.; Thiele, K.; Triviai, I.; Wieczorek, A.; Alawi, M.; Ohnezeit, D.; Kottlau, J.; Huang, J.; et al. Vertically transferred maternal immune cells promote neonatal immunity against early life infections. Nat. Commun. 2021, 12, 4706. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Khosrotehrani, K.; Way, S.S.; MacKenzie, T.C.; Bajema, I.; O’Donoghue, K. Forever Connected: The Lifelong Biological Consequences of Fetomaternal and Maternofetal Microchimerism. Clin. Chem. 2021, 67, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; El Haddad, M.; Donato, X.C.; Martin, G.V.; Bretelle, F.; Lesavre, N.; Cocallemen, J.F.; Martin, M.; Picard, C.; Albentosa, T.; et al. Grandmaternal cells in cord blood. eBioMedicine 2021, 74, 103721. [Google Scholar] [CrossRef] [PubMed]
- Yüzen, D.; Urbschat, C.; Schepanski, S.; Thiele, K.; Arck, P.C.; Mittrücker, H. Pregnancy-induced transfer of pathogen-specific T cells from mother to fetus in mice. EMBO Rep. 2023, 24, e56829. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Okada, A.; Sasano, K.; Endo, M.; Yamazaki, S.; Wang, X.; Shimbo, T.; Tomimatsu, T.; Kimura, T.; Tamai, K. Controlled induction of immune tolerance by mesenchymal stem cells transferred by maternal microchimerism. Biochem. Biophys. Res. Commun. 2021, 539, 83–88. [Google Scholar] [CrossRef]
- Papait, A.; Vertua, E.; Magatti, M.; Ceccariglia, S.; De Munari, S.; Silini, A.R.; Sheleg, M.; Ofir, R.; Parolini, O. Mesenchymal Stromal Cells from Fetal and Maternal Placenta Possess Key Similarities and Differences: Potential Implications for Their Applications in Regenerative Medicine. Cells 2020, 9, 127. [Google Scholar] [CrossRef]
- Zahavi-Goldstein, E.; Blumenfeld, M.; Fuchs-Telem, D.; Pinzur, L.; Rubin, S.; Aberman, Z.; Sher, N.; Ofir, R. Placenta-derived PLX-PAD mesenchymal-like stromal cells are efficacious in rescuing blood flow in hind limb ischemia mouse model by a dose- and site-dependent mechanism of action. Cytotherapy 2017, 19, 1438–1446. [Google Scholar] [CrossRef]
- Shirbaghaee, Z.; Hassani, M.; Heidari Keshel, S.; Soleimani, M. Emerging roles of mesenchymal stem cell therapy in patients with critical limb ischemia. Stem Cell Res. Ther. 2022, 13, 462. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, Y.; Zhang, Y.; Hao, G.; Liu, T.; Wang, L.; Yang, T.; Wang, Q.; Zhang, G.; Wei, J.; et al. Placental mesenchymal stem cells of fetal and maternal origins demonstrate different therapeutic potentials. Stem Cell Res. Ther. 2014, 5, 48. [Google Scholar] [CrossRef]
- Moonshi, S.S.; Adelnia, H.; Wu, Y.; Ta, H.T.; Moonshi, S.S.; Adelnia, H.; Wu, Y.; Ta, H.T.; Discipline, B. Placenta-Derived Mesenchymal Stem Cells for Treatment of Diseases: A Clinically Relevant Source. Adv. Ther. 2022, 5, 2200054. [Google Scholar] [CrossRef]
- de la Torre, P.; Flores, A.I. Current Status and Future Prospects of Perinatal Stem Cells. Genes 2021, 12, 6. [Google Scholar] [CrossRef]
- de la Torre, P.; Pérez-Lorenzo, M.J.; Alcázar-Garrido, Á.; Flores, A.I. Cell-Based Nanoparticles Delivery Systems for Targeted Cancer Therapy: Lessons from Anti-Angiogenesis Treatments. Molecules 2020, 25, 715. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Hua, J.; Liu, J.; Zhang, B.; Wang, W.; Yu, X.; Xu, J. Mesenchymal Stromal Cell-Based Targeted Therapy Pancreatic Cancer: Progress and Challenges. Int. J. Mol. Sci. 2023, 24, 3559. [Google Scholar] [CrossRef] [PubMed]
- Stene, L.C.; Gale, E.A.M. The prenatal environment and type 1 diabetes. Diabetologia 2013, 56, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Zorena, K.; Michalska, M.; Kurpas, M.; Jaskulak, M.; Murawska, A.; Rostami, S. Environmental Factors and the Risk of Developing Type 1 Diabetes—Old Disease and New Data. Biology 2022, 11, 608. [Google Scholar] [CrossRef]
- Riaz, F.; Wei, P.; Pan, F. PPARs at the crossroads of T cell differentiation and type 1 diabetes. Front. Immunol. 2023, 14, 1292238. [Google Scholar] [CrossRef]
- Darwiche, R.; Chong, M.M.W.; Santamaria, P.; Thomas, H.E.; Kay, T.W.H. Fas is detectable on beta cells in accelerated, but not spontaneous, diabetes in nonobese diabetic mice. J. Immunol. 2003, 170, 6292–6297. [Google Scholar] [CrossRef]
- Yolcu, E.S.; Shirwan, H.; Askenasy, N. Fas/Fas-Ligand Interaction As a Mechanism of Immune Homeostasis and β-Cell Cytotoxicity: Enforcement Rather Than Neutralization for Treatment of Type 1 Diabetes. Front. Immunol. 2017, 8, 342. [Google Scholar] [CrossRef]
- Nelson, J.L.; Gillespie, K.M.; Lambert, N.C.; Stevens, A.M.; Loubiere, L.S.; Rutledge, J.C.; Leisenring, W.M.; Erickson, T.D.; Yan, Z.; Mullarkey, M.E.; et al. Maternal microchimerism in peripheral blood in type 1 diabetes and pancreatic islet beta cell microchimerism. Proc. Natl. Acad. Sci. USA 2007, 104, 1637–1642. [Google Scholar] [CrossRef]
- vanZyl, B.; Planas, R.; Ye, Y.; Foulis, A.; de Krijger, R.R.; Vives-Pi, M.; Gillespie, K.M. Why are levels of maternal microchimerism higher in type 1 diabetes pancreas? Chimerism 2010, 1, 45–50. [Google Scholar] [CrossRef]
- Heninger, A.K.; Monti, P.; Wilhelm, C.; Schwaiger, P.; Kuehn, D.; Ziegler, A.G.; Bonifacio, E. Activation of islet autoreactive naïve T cells in infants is influenced by homeostatic mechanisms and antigen-presenting capacity. Diabetes 2013, 62, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- Tapia, G.; Mortimer, G.; Ye, J.; Gillard, B.T.; Chipper-Keating, S.; Mårild, K.; Viken, M.K.; Lie, B.A.; Joner, G.; Skrivarhaug, T.; et al. Maternal microchimerism in cord blood and risk of childhood-onset type 1 diabetes. Pediatr. Diabetes 2019, 20, 728–735. [Google Scholar] [CrossRef]
- Ushijima, K.; Okuno, M.; Ayabe, T.; Kikuchi, N.; Kawamura, T.; Urakami, T.; Yokota, I.; Amemiya, S.; Uchiyama, T.; Kikuchi, T.; et al. Low prevalence of maternal microchimerism in peripheral blood of Japanese children with type 1 diabetes. Diabet. Med. 2020, 37, 2131–2135. [Google Scholar] [CrossRef] [PubMed]
- Pagnini, I.; Vitale, A.; Selmi, C.; Cimaz, R.; Cantarini, L. Idiopathic Inflammatory Myopathies: An Update on Classification and Treatment with Special Focus on Juvenile Forms. Clin. Rev. Allergy Immunol. 2017, 52, 34–44. [Google Scholar] [CrossRef]
- Artlett, C.M.; Miller, F.W.; Rider, L.G. Persistent maternally derived peripheral microchimerism is associated with the juvenile idiopathic inflammatory myopathies. Rheumatology 2001, 40, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Arlett, C.M.; Ramos, R.; Jiminez, S.A.; Patterson, K.; Miller, F.W.; Rider, L.G.; Adams, B.; Borzy, M.; Cawkwell, G.; De Chadarevian, J.P.; et al. Chimeric cells of maternal origin in juvenile idiopathic inflammatory myopathies. Lancet 2000, 356, 2155–2156. [Google Scholar] [CrossRef]
- Reed, A.M.; Harwood, A.; Picornell, Y.J.; Kredich, D.W. Chimerism in children with juvenile dermatomyositis. Lancet 2000, 356, 2156–2157. [Google Scholar] [CrossRef]
- Ye, Y.; Van Zyl, B.; Varsani, H.; Wedderburn, L.R.; Ramanan, A.; Gillespie, K.M. Maternal microchimerism in muscle biopsies from children with juvenile dermatomyositis. Rheumatology 2012, 51, 987–991. [Google Scholar] [CrossRef]
- Artlett, C.M.; Sassi-Gaha, S.; Ramos, R.C.; Miller, F.W.; Rider, L.G. Chimeric cells of maternal origin do not appear to be pathogenic in the juvenile idiopathic inflammatory myopathies or muscular dystrophy. Arthritis Res. Ther. 2015, 17, 238. [Google Scholar] [CrossRef]
- Derdulska, J.M.; Rudnicka, L.; Szykut-Badaczewska, A.; Mehrholz, D.; Nowicki, R.J.; Barańska-Rybak, W.; Wilkowska, A. Neonatal lupus erythematosus—Practical guidelines. J. Perinat. Med. 2021, 49, 529–538. [Google Scholar] [CrossRef]
- Stevens, A.M.; Hermes, H.M.; Rutledge, J.C.; Buyon, J.P.; Nelson, J.L. Myocardial-tissue-specific phenotype of maternal microchimerism in neonatal lupus congenital heart block. Lancet 2003, 362, 1617–1623. [Google Scholar] [CrossRef]
- Stevens, A.M.; Hermes, H.M.; Lambert, N.C.; Nelson, J.L.; Meroni, P.L.; Cimaz, R. Maternal and sibling microchimerism in twins and triplets discordant for neonatal lupus syndrome-congenital heart block. Rheumatology 2005, 44, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Buyon, J.P.; Rupel, A.; Clancy, R.M. Congenital heart block: Do fetal factors fuel the fire from inflammation to fibrosis? Lupus 2003, 12, 731–734. [Google Scholar] [CrossRef]
- Kremer Hovinga, I.C.L.; Koopmans, M.; de Heer, E.; Bruijn, J.A.; Bajema, I.M. Chimerism in systemic lupus erythematosus-three hypotheses. Rheumatology 2007, 46, 200–208. [Google Scholar] [CrossRef]
- Kremer Hovinga, I.C.L.; Koopmans, M.; Baelde, H.J.; De Heer, E.; Bruijn, J.A.; Bajema, I.M. Tissue chimerism in systemic lupus erythematosus is related to injury. Ann. Rheum. Dis. 2007, 66, 1568–1573. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, M.; Okayama, A.; Nakamura, S.; Sasaki, T.; Murai, K.; Shiba, R.; Shinohara, M.; Tsubouchi, H. Detection of maternal-fetal microchimerism in the inflammatory lesions of patients with Sjögren’s syndrome. Ann. Rheum. Dis. 2002, 61, 1041–1046. [Google Scholar] [CrossRef]
- Adams, K.M.; Nelson, J.L. Microchimerism: An investigative frontier in autoimmunity and transplantation. JAMA 2004, 291, 1127–1131. [Google Scholar] [CrossRef]
- Muraji, T. Maternal microchimerism in biliary atresia: Are maternal cells effector cells, targets, or just bystanders? Chimerism 2014, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.F.N.; Atkins, C.J.; Naysmith, D.; Van Der Westhuizen, N.; Woo, J.; Nelson, J.L. Microchimerism in the rheumatoid nodules of rheumatoid arthritis patients. Arthritis Rheum. 2012, 64, 380–388. [Google Scholar] [CrossRef]
- Tapia, G.; Mortimer, G.; Ye, J.; Mårild, K.; Chipper-Keating, S.; Gillard, B.T.; Viken, M.K.; Lie, B.A.; Stene, L.C.; Gillespie, K.M.; et al. Maternal Microchimerism in Cord Blood and Risk of Celiac Disease in Childhood. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 321–327. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mpakosi, A.; Sokou, R.; Theodoraki, M.; Iacovidou, N.; Cholevas, V.; Kaliouli-Antonopoulou, C. Deciphering the Role of Maternal Microchimerism in Offspring Autoimmunity: A Narrative Review. Medicina 2024, 60, 1457. https://doi.org/10.3390/medicina60091457
Mpakosi A, Sokou R, Theodoraki M, Iacovidou N, Cholevas V, Kaliouli-Antonopoulou C. Deciphering the Role of Maternal Microchimerism in Offspring Autoimmunity: A Narrative Review. Medicina. 2024; 60(9):1457. https://doi.org/10.3390/medicina60091457
Chicago/Turabian StyleMpakosi, Alexandra, Rozeta Sokou, Martha Theodoraki, Nicoletta Iacovidou, Vasileios Cholevas, and Christiana Kaliouli-Antonopoulou. 2024. "Deciphering the Role of Maternal Microchimerism in Offspring Autoimmunity: A Narrative Review" Medicina 60, no. 9: 1457. https://doi.org/10.3390/medicina60091457