Chemical Effect of Bisphenol A on Non-Alcoholic Fatty Liver Disease

 ,
, 

 ,
, 



Abstract
:1. Introduction
The Exposition and Pharmacokinetics of BPA
2. EDCs and Metabolic Diseases
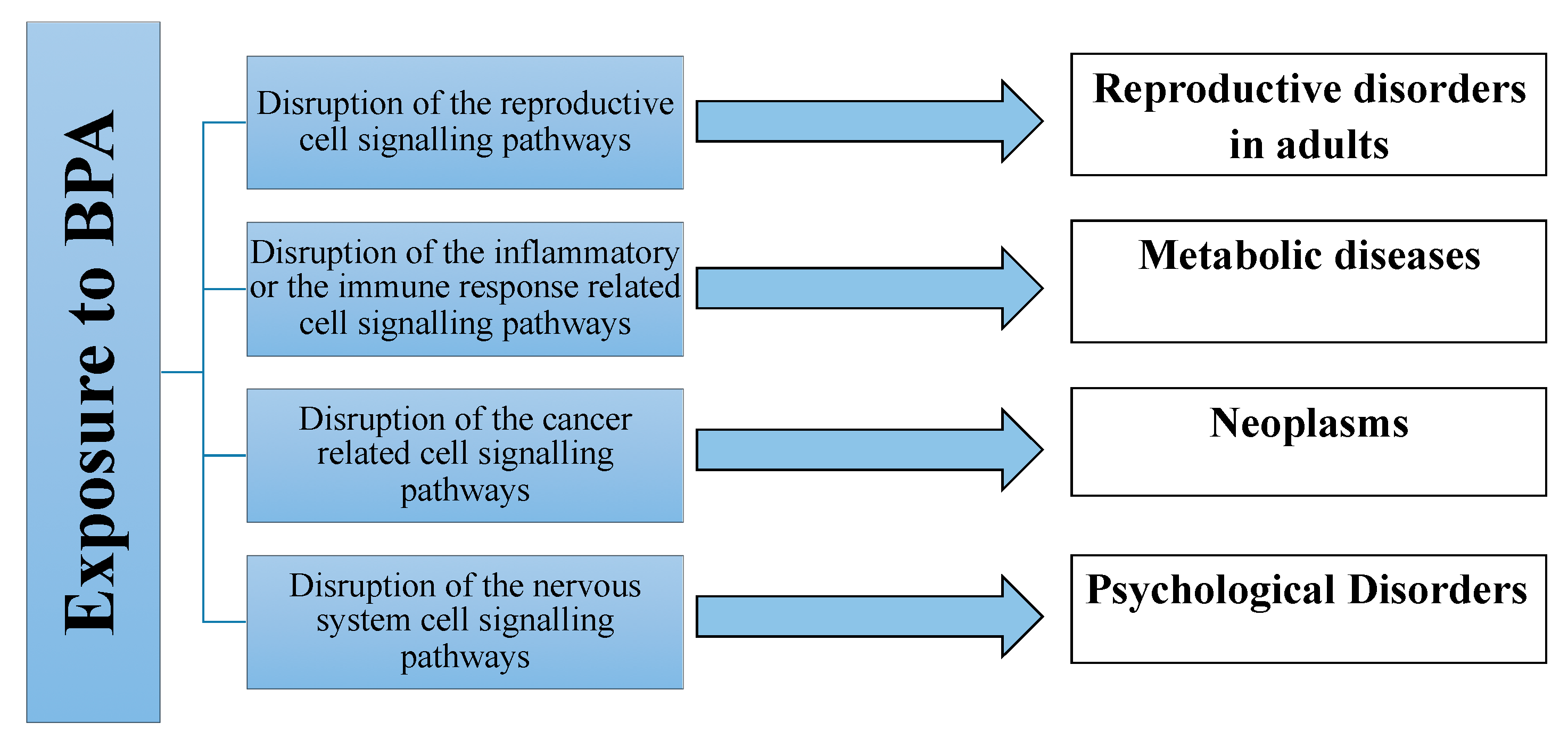
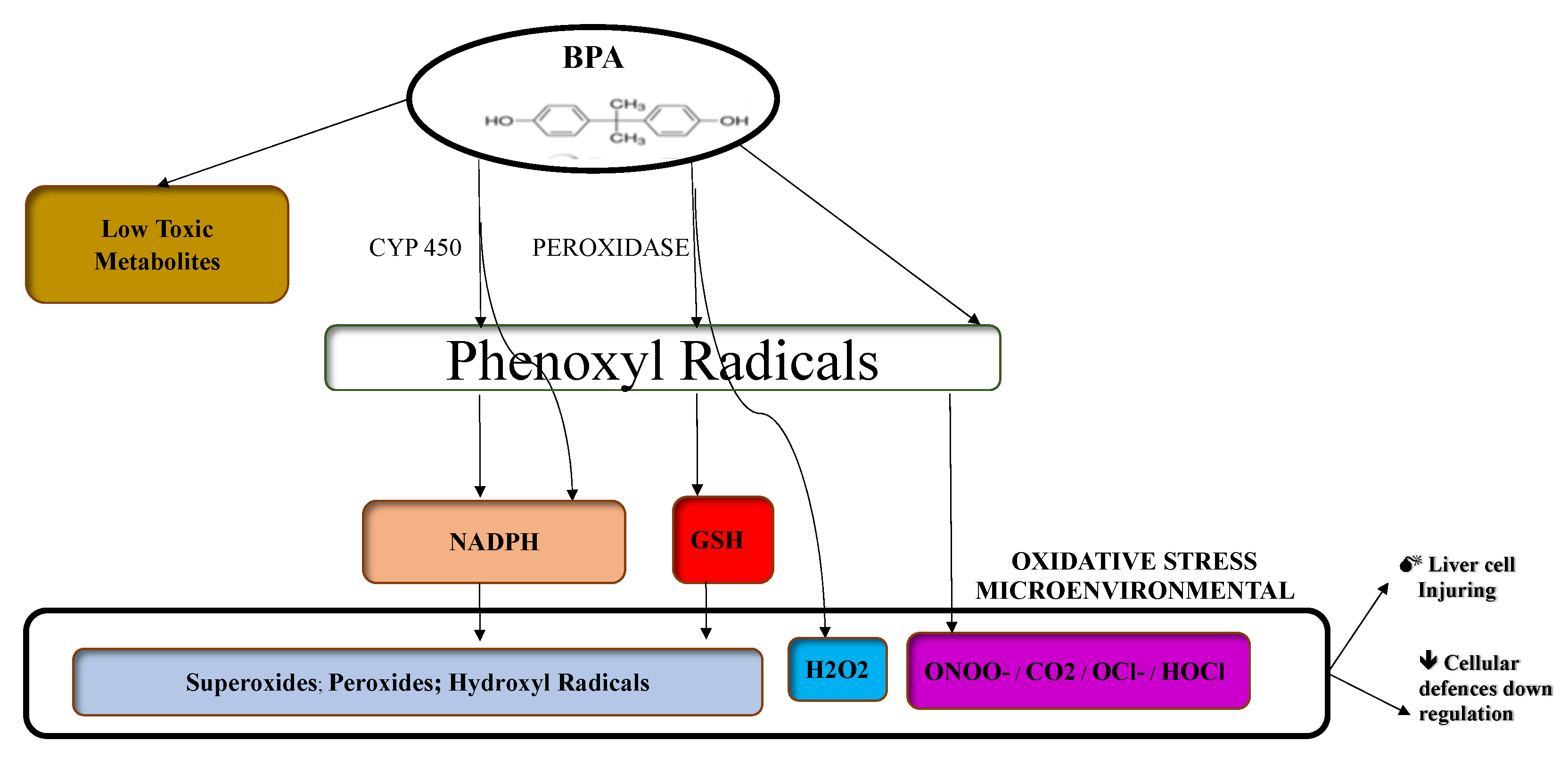
Mechanisms of Biological Action of BPA
3. NAFLD and EDCs
3.1. Hepatic TG Accumulation
3.2. Inflammation and Oxidative Stress: NAFLD-NASH Progression
3.3. NASH/HCC Progression
4. Conclusions
Author Contributions
Conflicts of Interest
List of Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| DCs | Endocrine disruptors |
| BPA | Bisphenol A |
| T2DM | Type 2 diabetes mellitus |
| PM | Particulate matter |
| DDT | Dichlorodiphenyltrichloroethane |
| TF | Tolylfluanid |
| POPs | Persistent organic pollutants |
| US | United States |
| EPA | Environmental protection agency |
| BW | Body weight |
| IR | Insulin Resistance |
| PCOs | Poly-cystic ovary syndrome |
| HDL | High-density lipoprotein |
| O-LDL | Oxidized low-density lipoproteins |
| LDLc | LDL cholesterol |
| IRS-1 | Insulin receptor substrate-1 |
| Akt2 | Protein kinase B-2 |
| ERα/β | Estrogen receptor α/β |
| AR | Androgen receptor |
| GPER | G protein-coupled estrogen receptor |
| GPR30 | G protein-coupled receptor 30 |
| IGF-1R | Insulin-like growth factor-1 receptor |
| ERRγ | Estrogen-related receptor gamma |
| MAPK | Mitogen-activated protein kinase |
| PI3K | Phosphatidylinositol-3 kinase |
| EGFR | Epidermal growth factor receptor |
| TNF-α | Tumor necrosis factor alpha |
| IL | Interleukin |
| GLUT | Glucose transporters |
| ROS | Reactive oxygen species |
| CAT | Catalase |
| MDA | Malondialdehyde |
| GSH | Glutathione |
| SOD | Superoxide dismutase |
| MiR | miRNAs |
| GAL | G-alanine and g-alanine-like peptide |
| CART | Cocaine- and amphetamine-regulated transcript |
| CGRP | Calcitonin Gene Related Peptide |
| 2AG | 2-arachidonoylglycerol |
| AEA | Anandamide |
| PEA | Palmitoylethanolamide |
| CB1 | Endocannabinoid receptor 1 |
| TG | Triglyceride |
| NASH | Steatohepatitis |
| HCC | Hepatocellular carcinoma |
| DNL | De novo lipogenesis |
| FFA | free fatty acids |
| SERBF1 | Sterol regulatory element-binding protein |
| UTR | 3’ untranslated region |
| M1KCs | Pro-inflammatory M1 phenotype |
| ORO | Oil red O |
| TBARS | Thiobarbituric acid reactive substances |
| PAMPs | Pathogens Associated Molecular Patterns |
| SCFA | Short Chain Fatty Acids |
| LPS | Lipopolysaccharides |
| γH2AX | Histon Family Member X |
| CYP1A1 | Cytochrome P450, family 1, subfamily A, polypeptide 1 |
| UGT1A1 | UDP-glucuronosyltransferase 1-1 |
| GSTA1 | Glutathione S-transferase A1 |
| GCLC | Glutamate–cysteine ligase catalytic subunit |
| GPX1 | Glutathione peroxidase 1 |
| GSR | Glutathione-disulfide reductase |
| SOD1A | Superoxide dismutase 1A |
| TP53 | Tumor Protein 53 |
| MDM2 | Mouse double minute 2 homolog |
| CDKN1A | Cyclin-dependent kinase inhibitor 1 |
| ERK | Extracellular signal-regulated kinase |
References
- Neuschwander-Tetri, B.A. Non-alcoholic fatty liver disease. BMC Med. 2017, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, W.R. Nonobese Fatty Liver Disease. Clinical gastroenterology and hepatology: The official clinical practice. J. Am. Gastroenterol. Assoc. 2017, 15, 474–485. [Google Scholar]
- Nicolucci, C.; Errico, S.; Federico, A.; Dallio, M.; Loguercio, C.; Diano, N. Human exposure to Bisphenol A and liver health status: Quantification of urinary and circulating levels by LC–MS/MS. J. Pharm. Biomed. Anal. 2017, 140, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxidative Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Masarone, M.; Persico, M.; Loguercio, C. The epidemiology of non-alcoholic fatty liver disease and its connection with cardiovascular disease: Role of endothelial dysfunction. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4731–4741. [Google Scholar] [PubMed]
- Sherlock, S. Sherlock’s Diseases of the Liver and Biliary System, 13th ed.; Dooley, J.S., Garcia-Tsao, G., Pinzani, M., Eds.; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Yarana, C.; Thompson, H.; Chaiswing, L.; Butterfield, D.A.; Weiss, H.; Bondada, S.; AlHakeem, S.; Sukati, S.; Clair, D.K.S. Extracellular vesicle-mediated macrophage activation: An insight into the mechanism of thioredoxin-mediated immune activation. Redox Biol. 2019, 26, 101237. [Google Scholar] [CrossRef]
- Abdeen, A.; Abou-Zaid, O.A.; Abdel-Maksoud, H.A.; Aboubakr, M.; Abdelkader, A.; AbdelNaby, A.; Abo-Ahmed, A.I.; El-Mleeh, A.; Mostafa, O.; Abdel-Daim, M.; et al. Cadmium overload modulates piroxicam-regulated oxidative damage and apoptotic pathways. Environ. Sci. Pollut. Res. 2019, 26, 25167–25177. [Google Scholar] [CrossRef] [PubMed]
- Sharp, K.P.H.; Schultz, M.; Coppell, K.J. Is non-alcoholic fatty liver disease a reflection of what we eat or simply how much we eat? JGH Open 2018, 2, 59–74. [Google Scholar] [CrossRef]
- Kirkley, A.G.; Sargis, R.M. Environmental Endocrine Disruption of Energy Metabolism and Cardiovascular Risk. Curr. Diabetes Rep. 2014, 14, 494. [Google Scholar] [CrossRef]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef]
- Lama, S.; Vanacore, D.; Diano, N.; Nicolucci, C.; Errico, S.; Dallio, M.; Federico, A.; Loguercio, C.; Stiuso, P. Ameliorative effect of Silybin on bisphenol A induced oxidative stress, cell proliferation and steroid hormones oxidation in HepG2 cell cultures. Sci. Rep. 2019, 9, 3228. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Deretzi, G.; Zavos, C.; Mantzoros, C.S. The emerging role of endocrine disruptors in pathogenesis of insulin resistance: A concept implicating nonalcoholic fatty liver disease. Curr. Mol. Med. 2012, 12, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Bourguignon, J.P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Le Magueresse-Battistoni, B.; Labaronne, E.; Vidal, H.; Naville, D. Endocrine disrupting chemicals in mixture and obesity, diabetes and related metabolic disorders. World J. Biol. Chem. 2017, 8, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Signorile, P.G.; Pietro, G.S. Endocrine disruptors in utero cause ovarian damages linked to endometriosis. Front. Biosci. 2012, 4, 1724–1730. [Google Scholar] [CrossRef]
- Thoene, M.; Rytel, L.; Dzika, E.; Włodarczyk, A.; Kruminis-Kaszkiel, E.; Konrad, P.; Wojtkiewicz, J. Bisphenol A Causes Liver Damage and Selectively Alters the Neurochemical Coding of Intrahepatic Parasympathetic Nerves in Juvenile Porcine Models under Physiological Conditions. Int. J. Mol. Sci. 2017, 18, 2726. [Google Scholar] [CrossRef] [PubMed]
- Malaisé, Y.; Menard, S.; Cartier, C.; Gaultier, E.; Lasserre, F.; Lencina, C.; Harkat, C.; Geoffre, N.; Lakhal, L.; Castan, I.; et al. Gut dysbiosis and impairment of immune system homeostasis in perinatally-exposed mice to Bisphenol A precede obese phenotype development. Sci. Rep. 2017, 7, 14472. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Kang, J.H. Bisphenol A (BPA) and cell signaling pathways. Biotechnol. Adv. 2018, 36, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. (Elmsford NY.) 2007, 24, 139–177. [Google Scholar] [CrossRef]
- Muñoz-De-Toro, M.; Markey, C.M.; Wadia, P.R.; Luque, E.H.; Rubin, B.S.; Sonnenschein, C.; Soto, A.M. Perinatal Exposure to Bisphenol-A Alters Peripubertal Mammary Gland Development in Mice. Endocrinology 2005, 146, 4138–4147. [Google Scholar] [CrossRef]
- Mikołajewska, K.; Stragierowicz, J.; Gromadzińska, J. Bisphenol, A—Application, sources of exposure and potential risks in infants, children and pregnant women. Int. J. Occup. Med. Environ. Health 2015, 28, 209–241. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Khan, A.; Jee, S.H.; Lee, H.S.; Hwang, M.S.; Koo, Y.E.; Park, Y.H. High resolution metabolomics to determines the risk associated with bisphenol A exposure in humans. Environ. Toxicol. Pharmacol. 2018, 58, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dallio, M.; Masarone, M.; Errico, S.; Gravina, A.G.; Nicolucci, C.; Di Sarno, R.; Gionti, L.; Tuccillo, C.; Persico, M.; Stiuso, P.; et al. Role of bisphenol A as environmental factor in the promotion of non-alcoholic fatty liver disease: In vitro and clinical study. Aliment. Pharmacol. Ther. 2018, 47, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. (Elmsford, NY) 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Sargis, R.M.; Neel, B.A.; Brock, C.O.; Lin, Y.; Hickey, A.T.; Carlton, D.A.; Brady, M.J. The Novel Endocrine Disruptor Tolylfluanid Impairs Insulin Signaling in Primary Rodent and Human Adipocytes through a Reduction in Insulin Receptor Substrate-1 Levels. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.M. Early-life exposure to EDCs: Role in childhood obesity and neurodevelopment. Nature reviews. Endocrinology 2017, 13, 161–173. [Google Scholar] [PubMed]
- Chevalier, N.; Fénichel, P. Bisphenol A: Targeting metabolic tissues. Rev. Endocr. Metab. Disord. 2015, 16, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Terasaka, S.; Kiyama, R. Bisphenol A induces a rapid activation of Erk1/2 through GPR30 in human breast cancer cells. Environ. Pollut. (Barking Essex 1987) 2011, 159, 212–218. [Google Scholar] [CrossRef]
- Wetherill, Y.B.; Petre, E.C.; Monk, K.R.; Puga, A.; Knudsen, E.K. The xenoestrogen bisphenol A induces inappropriate androgen receptor activation and mitogenesis in prostatic adenocarcinoma cells. Mol. Cancer Ther. 2002, 1, 515–524. [Google Scholar]
- Pupo, M.; Pisano, A.; Lappano, R.; Santolla, M.F.; De Francesco, E.M.; Abonante, S.; Rosano, C.; Maggiolini, M. Bisphenol A Induces Gene Expression Changes and Proliferative Effects through GPER in Breast Cancer Cells and Cancer-Associated Fibroblasts. Environ. Health Perspect. 2012, 120, 1177–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, K.A.; Park, M.A.; Kang, N.H.; Yi, B.R.; Hyun, S.H.; Jeung, E.B.; Choi, K.C. Anticancer effect of genistein on BG-1 ovarian cancer growth induced by 17 beta-estradiol or bisphenol A via the suppression of the crosstalk between estrogen receptor alpha and insulin-like growth factor-1 receptor signaling pathways. Toxicol. Appl. Pharmacol. 2013, 272, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.S.; Chen, H.Q.; Chen, Y.S.; Qiu, K.F.; Zheng, X.B.; Li, G.C.; Yang, H.D.; Wen, C.J. Bisphenol A stimulates human lung cancer cell migration via upregulation of matrix metalloproteinases by GPER/EGFR/ERK1/2 signal pathway. Biomed. Pharmacother. 2014, 68, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Alonso-Magdalena, P.; Garcia-Arevalo, M.; Novials, A.; Muhammed, S.J.; Salehi, A.; Gustafsson, J.A.; Quesada, I.; Nadal, A. Rapid insulinotropic action of low doses of bisphenol-A on mouse and human islets of Langerhans: Role of estrogen receptor beta. PLoS ONE 2012, 7, e31109. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Yang, H.; Ahn, C.; Kang, H.Y.; Hong, E.J.; Jaung, E.B. Effects of xenoestrogens on streptozotocin-induced diabetic mice. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2014, 65, 273–282. [Google Scholar]
- Gassman, N.R. Induction of oxidative stress by bisphenol A and its pleiotropic effects. Environ. Mol. Mutagen. 2017, 58, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, X.; Liu, Z.; Li, Y.; Yao, D.; Sun, L.; Wang, B.; Ma, Y.; Wang, L.; Zhang, Y. Investigation of the effect for bisphenol A on oxidative stress in human hepatocytes and its interaction with catalase. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 221, 117149. [Google Scholar] [CrossRef]
- Hassani, F.V.; Abnous, K.; Mehri, S.; Jafarian, A.; Birner-Gruenberger, R.; Robati, R.Y.; Hosseinzadeh, H. Proteomics and phosphoproteomics analysis of liver in male rats exposed to bisphenol A: Mechanism of hepatotoxicity and biomarker discovery. Food Chem. Toxicol. 2018, 112, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Acaroz, U.; Ince, S.; Arslan-Acaroz, D.; Gurler, Z.; Demirel, H.H.; Kucukkurt, I.; Eryavuz, A.; Kara, R.; Varol, N.; Zhu, K. Bisphenol-A induced oxidative stress, inflammatory gene expression, and metabolic and histopathological changes in male Wistar albino rats: Protective role of boron. Toxicol. Res. 2019, 8, 262–269. [Google Scholar] [CrossRef]
- Martella, A.; Silvestri, C.; Maradonna, F.; Gioacchini, G.; Allarà, M.; Radaelli, G.; Overby, D.R.; Di Marzo, V.; Carnevali, O. Bisphenol A Induces Fatty Liver by an Endocannabinoid-Mediated Positive Feedback Loop. Endocrinology 2016, 157, 1751–1763. [Google Scholar] [CrossRef] [Green Version]
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease-An Evolving View. Clin. Liver Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Marmugi, A.; Ducheix, M.S.; Lasserre, M.F.; Polizzi, M.; Paris, A.; Priymenko, N.; Bertrand-Michel, J.; Pineau, T.; Guillou, H.; Martin, P.G.; et al. Low doses of Bisphenol A induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver. Ann. Endocrinol. 2012, 73, 420. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Rafiq, N.; Henry, L.; Loomba, R.; Makhlouf, H.; Goodman, Z. Nonalcoholic steatofibrosis independently predicts mortality in nonalcoholic fatty liver disease. Hepatol. Commun. 2017, 1, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The histological course of nonalcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar] [CrossRef]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology (Baltimore, Md.) 2006, 44, 865–873. [Google Scholar] [CrossRef]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.-X.; Lezmi, S. Developmental Bisphenol A (BPA) Exposure Leads to Sex-specific Modification of Hepatic Gene Expression and Epigenome at Birth that May Exacerbate High-fat Diet-induced Hepatic Steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Rametta, R.; Dongiovanni, P.; Maggioni, M.; Fracanzani, A.L.; Zappa, M.; Lattuada, E.; Roviaro, G.; Fargion, S. Increased Expression and Activity of the Transcription Factor FOXO1 in Nonalcoholic Steatohepatitis. Diabetes 2008, 57, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef]
- Adams, L.A.; Angulo, P.; Lindor, K.D. Nonalcoholic fatty liver disease. Can. Med. Assoc. J. 2005, 172, 899–905. [Google Scholar] [CrossRef] [Green Version]
- Dunder, L.; Lejonklou, M.H.; Lind, L.; Risérus, U.; Lind, P.M. Low-dose developmental bisphenol A exposure alters fatty acid metabolism in Fischer 344 rat offspring. Environ. Res. 2018, 166, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Labaronne, E.; Pinteur, C.; Vega, N.; Pesenti, S.; Julien, B.; Meugnier-Fouilloux, E.; Vidal, H.; Naville, D.; Le Magueresse-Battistoni, B. Low-dose pollutant mixture triggers metabolic disturbances in female mice leading to common and specific features as compared to a high-fat diet. J. Nutr. Biochem. 2017, 45, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ding, D.; Huang, Q.; Liu, Q.; Lu, H.; Lu, Y.; Chi, Y.; Sun, X.; Ye, G.; Zhu, H.; et al. Downregulation of miR-192 causes hepatic steatosis and lipid accumulation by inducing SREBF1: Novel mechanism for bisphenol A-triggered non-alcoholic fatty liver disease. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2017, 1862, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Huc, L.; Lemarie, A.; Gueraud, F.; Helies-Toussaint, C. Low concentrations of bisphenol A induce lipid accumulation mediated by the production of reactive oxygen species in the mitochondria of HepG2 cells. Toxicol. Vitr. 2012, 26, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nature reviews. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar]
- Stiuso, P.; Scognamiglio, I.; Murolo, M.; Ferranti, P.; De Simone, C.; Rizzo, M.R.; Tuccillo, C.; Caraglia, M.; Loguercio, C.; Federico, A. Serum Oxidative Stress Markers and Lipidomic Profile to Detect NASH Patients Responsive to an Antioxidant Treatment: A Pilot Study. Oxidative Med. Cell. Longev. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Federico, A.; Conti, V.; Russomanno, G.; Dallio, M.; Masarone, M.; Stiuso, P.; Tuccillo, C.; Caraglia, M.; Manzo, V.; Persico, M.; et al. A Long-term Treatment with Silybin in Patients with Non-alcoholic Steatohepatitis Stimulates Catalase Activity in Human Endothelial Cells. Vivo 2017, 31, 609–618. [Google Scholar] [Green Version]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate immunity and inflammation in NAFLD/NASH. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef]
- Alisi, A.; Carpino, G.; Oliveira, F.L.; Panera, N.; Nobili, V.; Gaudio, E. The Role of Tissue Macrophage-Mediated Inflammation on NAFLD Pathogenesis and Its Clinical Implications. Mediat. Inflamm. 2017, 2017, 1–15. [Google Scholar] [CrossRef]
- Reddivari, L.; Veeramachaneni, D.N.R.; Walters, W.A.; Lozupone, C.; Palmer, J.; Hewage, M.K.K.; Bhatnagar, R.; Amir, A.; Kennett, M.J.; Knight, R.; et al. Perinatal Bisphenol a Exposure Induces Chronic Inflammation in Rabbit Offspring via Modulation of Gut Bacteria and Their Metabolites. mSystems 2017, 2, e00093-17. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Caprio, G.G.; Ormando, V.M.; Loguercio, C. Gut microbiota and gastrointestinal tract, liver and pancreas: From physiology to pathology. Minerva Gastroenterol. Dietol. 2017, 63, 385–398. [Google Scholar] [PubMed]
- Braniste, V.; Jouault, A.; Gaultier, E.; Polizzi, A.; Buisson-Brenac, C.; Leveque, M.; Martin, P.G.; Theodorou, V.; Fioramonti, J.; Houdeau, E. Impact of oral bisphenol A at reference doses on intestinal barrier function and sex differences after perinatal exposure in rats. Proc. Natl. Acad. Sci. USA 2010, 107, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Dallio, M.; Godos, J.; Loguercio, C.; Salomone, F.; Loguercio, C. Targeting gut-liver axis for the treatment of nonalcoholic steatohepatitis: Translational and clinical evidence. Transl. Res. 2016, 167, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Kolsek, K.; Mavri, J.; Sollner Dolenc, M. Reactivity of bisphenol A-3,4-quinone with DNA. A quantum chemical study. Toxicol. Vitr. 2012, 26, 102–106. [Google Scholar] [CrossRef]
- Tsutsui, T.; Tamura, Y.; Yagi, E.; Hasegawa, K.; Takahashi, M.; Maizumi, N.; Yamaguchi, F.; Barrett, J.C. Bisphenol-A induces cellular transformation, aneuploidy and DNA adduct formation in cultured Syrian hamster embryo cells. Int. J. Cancer 1998, 75, 290–294. [Google Scholar] [CrossRef]
- Xin, L.; Lin, Y.; Wang, A.; Zhu, W.; Liang, Y.; Su, X.; Hong, C.; Wan, J.; Wang, Y.; Tian, H. Cytogenetic evaluation for the genotoxicity of bisphenol-A in Chinese hamster ovary cells. Environ. Toxicol. Pharmacol. 2015, 40, 524–529. [Google Scholar] [CrossRef]
- Izzotti, A.; Kanitz, S.; D’Agostini, F.; Camoirano, A.; De Flora, S. Formation of adducts by bisphenol A, an endocrine disruptor, in DNA in vitro and in liver and mammary tissue of mice. Mutat. Res. Toxicol. Environ. Mutagen. 2009, 679, 28–32. [Google Scholar] [CrossRef]
- Fic, A.; Zegura, B.; Sollner Dolenc, M.; Filipic, M.; Peterlin Masic, L. Mutagenicity and DNA damage of bisphenol A and its structural analogues in HepG2 cells. Arh. Hig. Rada Toksikol. 2013, 64, 189–200. [Google Scholar] [CrossRef]
- Hercog, K.; Maisanaba, S.; Filipič, M.; Sollner-Dolenc, M.; Kač, L.; Žegura, B. Genotoxic activity of bisphenol A and its analogues bisphenol S, bisphenol F and bisphenol AF and their mixtures in human hepatocellular carcinoma (HepG2) cells. Sci. Total Environ. 2019, 687, 267–276. [Google Scholar] [CrossRef]
- Weinhouse, C.; Anderson, O.S.; Bergin, I.L.; Vandenbergh, D.J.; Gyekis, J.P.; Dingman, M.A.; Yang, J.; Dolinoy, D.C. Dose-Dependent Incidence of Hepatic Tumors in Adult Mice following Perinatal Exposure to Bisphenol A. Environ. Health Perspect. 2014, 122, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Endocrine Disruptors | ||
|---|---|---|
| Arsenic | Insecticides | Fire retardants |
| Atrazine | Polychlorobiphenyl | Estradiol |
| Bisphenol A | Cadmium | Estrone |
| Lead | Parabens | Fungicides |
| Mercury | Pesticides | Perchlorate |
| Phytoestrogens | Bis (2-ethylhexyl)phthalate | Triclosan |
| Glycol ethers | Polycyclic aromatic hydrocarbons | Perfluorinated chemicals |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dallio, M.; Diano, N.; Masarone, M.; Gravina, A.G.; Patanè, V.; Romeo, M.; Di Sarno, R.; Errico, S.; Nicolucci, C.; Abenavoli, L.; et al. Chemical Effect of Bisphenol A on Non-Alcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 3134. https://doi.org/10.3390/ijerph16173134
Dallio M, Diano N, Masarone M, Gravina AG, Patanè V, Romeo M, Di Sarno R, Errico S, Nicolucci C, Abenavoli L, et al. Chemical Effect of Bisphenol A on Non-Alcoholic Fatty Liver Disease. International Journal of Environmental Research and Public Health. 2019; 16(17):3134. https://doi.org/10.3390/ijerph16173134
Chicago/Turabian StyleDallio, Marcello, Nadia Diano, Mario Masarone, Antonietta Gerarda Gravina, Vittorio Patanè, Mario Romeo, Rosa Di Sarno, Sonia Errico, Carla Nicolucci, Ludovico Abenavoli, and et al. 2019. "Chemical Effect of Bisphenol A on Non-Alcoholic Fatty Liver Disease" International Journal of Environmental Research and Public Health 16, no. 17: 3134. https://doi.org/10.3390/ijerph16173134
APA StyleDallio, M., Diano, N., Masarone, M., Gravina, A. G., Patanè, V., Romeo, M., Di Sarno, R., Errico, S., Nicolucci, C., Abenavoli, L., Scarpellini, E., Boccuto, L., Persico, M., Loguercio, C., & Federico, A. (2019). Chemical Effect of Bisphenol A on Non-Alcoholic Fatty Liver Disease. International Journal of Environmental Research and Public Health, 16(17), 3134. https://doi.org/10.3390/ijerph16173134