
The Sulfotransferase SULT1C2 Is Epigenetically Activated and Transcriptionally Induced by Tobacco Exposure and Is Associated with Patient Outcome in Lung Adenocarcinoma

 and
and
Abstract
:
1. Background
2. Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
2.4. Next-Generation Sequencing Analysis
2.5. The Cancer Genome Atlas (TCGA) Datasets
2.6. Survival Curves
2.7. Microarray Analysis
2.8. SULT1C2 Promoter Construction
2.9. In Vitro Methylation
2.10. Transfection and Luciferase Assay
2.11. Aza-CdR Treatment
2.12. MethyLight Assay
2.13. Chromatin Immunoprecipitation (ChIP) Assay
2.14. Statistical Analysis
3. Results
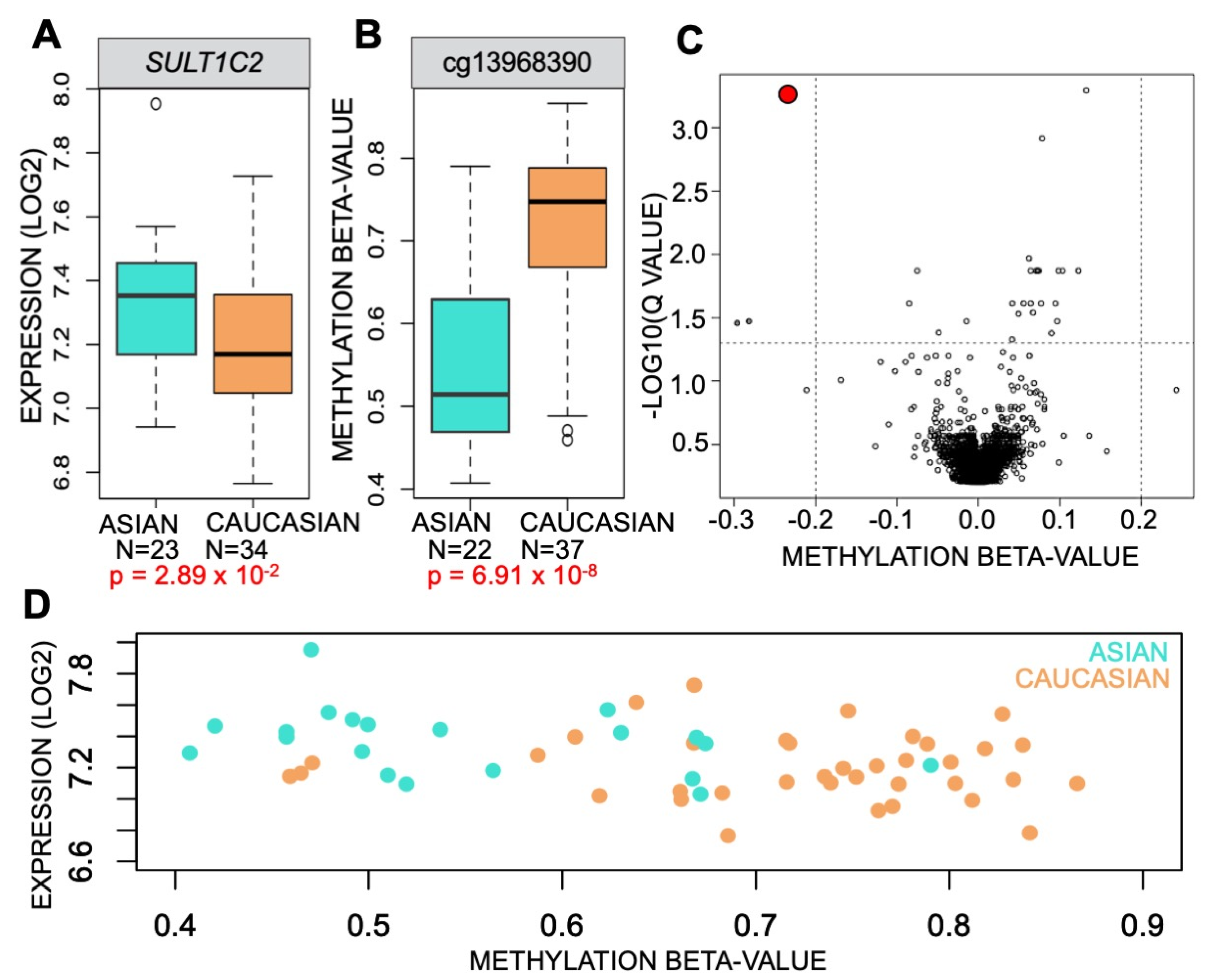
3.1. Methylation of SULT1C2 Promoter Is Altered in Human Lung
3.2. DNA Methylation Represses Transcription of SULT1C2 Promoter
3.3. CSC Alters AHR Occupancy at the SULT1C2 Promoter
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CSC | cigarette smoke condensate |
| LUAD | lung adenocarcinoma |
| AdjNTL | adjacent non-tumor lung |
| XRE | xenobiotic response element |
| AHR | aryl hydrocarbon receptor |
| PAH | polycyclic aromatic hydrocarbons |
| 5-aza-CdR | 5-Aza-deoxycytidine |
| ETS | environmental tobacco smoke |
| TSS | transcriptional start site |
| ALU | arthrobacter luteus repetitive element |
| RLU | relative light units |
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.P.; Tsai, M.H.; Lee, J.M.; Hsu, C.P.; Chen, P.C.; Lin, C.W.; Shih, J.-Y.; Yang, P.-C.; Hsiao, C.K.; Lai, L.-C.; et al. Identification of a novel biomarker, SEMA5A, for non-small cell lung carcinoma in nonsmoking women. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2590–2597. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.C.; Lee, C.H.; Chen, M.J.; Huang, C.C.; Chang, W.Y.; Lin, H.J.; Wang, H.Z.; Chang, P.Y. Risk factors for primary lung cancer among non-smoking women in Taiwan. Int. J. Epidemiol. 1997, 26, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovino, G.A.; Biener, L.; Hartman, A.M.; Marcus, S.E.; Schooley, M.W.; Pechacek, T.F.; Vallone, D. Monitoring the tobacco use epidemic I. Overview: Optimizing measurement to facilitate change. Prev. Med. 2009, 48, S4–S10. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, J.; Govindan, R. Lung cancer in ‘Never-smokers’: A unique entity. Oncology 2010, 24, 29–35. [Google Scholar] [PubMed]
- Ko, Y.C.; Cheng, L.S.; Lee, C.H.; Huang, J.J.; Huang, M.S.; Kao, E.L.; Wang, H.Z.; Lin, H.J. Chinese food cooking and lung cancer in women nonsmokers. Am. J. Epidemiol. 2000, 151, 140–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, T.A.; Pokhrel, P. Ethnic differences in smoking rate, nicotine dependence, and cessation-related variables among adult smokers in Hawaii. J. Community Health 2012, 37, 1226–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, F.; Lee, E.; Zheng, Y. Trajectory of smoking behavior change among Chinese immigrant smokers. PLoS ONE 2021, 16, e0246280. [Google Scholar] [CrossRef] [PubMed]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 2006, 90, 5–22. [Google Scholar] [CrossRef]
- Allali-Hassani, A.; Pan, P.W.; Dombrovski, L.; Najmanovich, R.; Tempel, W.; Dong, A.; Loppnau, P.; Martin, F.; Thornton, J.; Edwards, A.M.; et al. Structural and chemical profiling of the human cytosolic sulfotransferases. PLoS Biol. 2007, 5, e97. [Google Scholar]
- Weinshilboum, R.M.; Otterness, D.M.; Aksoy, I.A.; Wood, T.C.; Her, C.; Raftogianis, R.B. Sulfation and sulfotransferases 1: Sulfotransferase molecular biology: cDNAs and genes. FASEB J. 1997, 11, 3–14. [Google Scholar] [CrossRef]
- Yasuda, S.; Idell, S.; Fu, J.; Carter, G.; Snow, R.; Liu, M.C. Cigarette smoke toxicants as substrates and inhibitors for human cytosolic SULTs. Toxicol. Appl. Pharmacol. 2007, 221, 13–20. [Google Scholar] [CrossRef]
- Her, C.; Kaur, G.P.; Athwal, R.S.; Weinshilboum, R.M. Human sulfotransferase SULT1C1: cDNA cloning, tissue-specific expression, and chromosomal localization. Genomics 1997, 41, 467–470. [Google Scholar] [CrossRef]
- Rondini, E.A.; Fang, H.; Runge-Morris, M.; Kocarek, T.A. Regulation of human cytosolic sulfotransferases 1C2 and 1C3 by nuclear signaling pathways in LS180 colorectal adenocarcinoma cells. Drug Metab. Dispos. 2014, 42, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, S.S. Cigarette smoking and lung cancer: Chemical mechanisms and approaches to prevention. Lancet Oncol. 2002, 3, 461–469. [Google Scholar] [CrossRef]
- Doll, R. Cancers weakly related to smoking. Br. Med. Bull. 1996, 52, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Port, J.L.; Yamaguchi, K.; Du, B.; De Lorenzo, M.; Chang, M.; Heerdt, P.M.; Kopelovich, L.; Marcus, C.B.; Altorki, N.K.; Subbaramaiah, K.; et al. Tobacco smoke induces CYP1B1 in the aerodigestive tract. Carcinogenesis 2004, 25, 2275–2281. [Google Scholar] [CrossRef] [PubMed]
- Köhle, C.; Bock, K.W. Coordinate regulation of Phase I and II xenobiotic metabolisms by the Ah receptor and Nrf2. Biochem. Pharmacol. 2007, 73, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef]
- Dietrich, C. Antioxidant Functions of the Aryl Hydrocarbon Receptor. Stem Cells Int. 2016, 2016, 7943495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenas-Huertero, F.; Zaragoza-Ojeda, M.; Sánchez-Alarcón, J.; Milić, M.; Šegvić Klarić, M.; Montiel-González, J.M.; Valencia-Quintana, R. Involvement of Ahr Pathway in Toxicity of Aflatoxins and Other Mycotoxins. Front. Microbiol. 2019, 10, 2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papoutsis, A.J.; Selmin, O.I.; Borg, J.L.; Romagnolo, D.F. Gestational exposure to the AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: Preventive effects of resveratrol. Mol. Carcinog. 2015, 54, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Vorrink, S.U.; Hudachek, D.R.; Domann, F.E. Epigenetic determinants of CYP1A1 induction by the aryl hydrocarbon receptor agonist 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126). Int. J. Mol. Sci. 2014, 15, 13916–13931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef] [Green Version]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Li, R.; Mav, D.; Grimm, S.A.; Jothi, R.; Shah, R.; Wade, P.A. Fine-tuning of epigenetic regulation with respect to promoter CpG content in a cell type-specific manner. Epigenetics 2014, 9, 747–759. [Google Scholar] [CrossRef] [Green Version]
- Eckhardt, F.; Lewin, J.; Cortese, R.; Rakyan, V.K.; Attwood, J.; Burger, M.; Burton, J.; Cox, T.J.; Davies, R.; Down, T.A.; et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006, 38, 1378–1385. [Google Scholar] [CrossRef] [Green Version]
- Gal-Yam, E.N.; Egger, G.; Iniguez, L.; Holster, H.; Einarsson, S.; Zhang, X.; Lin, J.C.; Liang, G.; Jones, P.A.; Tanay, A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. USA 2008, 105, 12979–12984. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Shahabi, S.; Kumaran, V.; Castillo, J.; Cong, Z.; Nandagopal, G.; Mullen, D.J.; Alvarado, A.; Ramos Correa, M.; Saizan, A.; Goel, R.; et al. LINC00261 is an epigenetically-regulated tumor suppressor that is essential for activation of the DNA damage response. Cancer Res. 2019, 79, 3050–3062. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11.4.1–11.4.9. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.K.; Tripathi, P.; Koneva, L.A.; Cavalcante, R.G.; Craig, N.; Scruggs, A.M.; Sartor, M.A.; Deng, F.; Chen, Y. Effect of concentration and duration of particulate matter exposure on the transcriptome and DNA methylome of bronchial epithelial cells. Environ. Epigenet. 2021, 7, dvaa022. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; Eyring, K.; De, S.; Yang, I.V.; Schwartz, D.A. Fast and accurate alignment of long bisulfite-seq reads. arXiv 2014, arXiv:1401.1129. [Google Scholar]
- Suzuki, A.; Wakaguri, H.; Yamashita, R.; Kawano, S.; Tsuchihara, K.; Sugano, S.; Suzuki, Y.; Nakai, N. DBTSS as an integrative platform for transcriptome, epigenome and genome sequence variation data. Nucleic Acids Res. 2015, 43, D87–D91. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, S.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Selamat, S.A.; Chung, B.S.; Girard, L.; Zhang, W.; Zhang, Y.; Campan, M.; Siegmund, K.D.; Koss, M.N.; Hagen, J.A.; Lam, W.L.; et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012, 22, 1197–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [Green Version]
- Zong, F.Y.; Fu, X.; Wei, W.J.; Luo, Y.G.; Heiner, M.; Cao, L.J.; Fang, Z.; Fang, R.; Lu, D.; Ji, D.; et al. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014, 10, e1004289. [Google Scholar] [CrossRef] [PubMed]
- Wu, B. Substrate inhibition kinetics in drug metabolism reactions. Drug Metab. Rev. 2011, 43, 440–456. [Google Scholar] [CrossRef]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [Green Version]
- Tsaprouni, L.G.; Yang, T.P.; Bell, J.; Dick, K.J.; Kanoni, S.; Nisbet, J.; Viñuela, A.; Grundberg, E.; Nelson, C.P.; Meduri, E.; et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 2014, 9, 1382–1396. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Srivastava, S. The early detection research network: 10-year outlook. Clin. Chem. 2013, 59, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Klug, M.; Rehli, M. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics 2006, 1, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Cortez, C.C.; Yang, X.; Nichols, P.W.; Jones, P.A.; Liang, G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum. Mol. Genet. 2011, 20, 4299–4310. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Short, N.J.; Montalban-Bravo, G.; Huang, X.; Bueso-Ramos, C.; Qiao, W.; Yang, H.; Zhao, C.; Kadia, T.; Borthakur, G.; et al. Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN. Blood 2017, 130, 1514–1522. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012, 21, 430–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://portal.biobase-international.com (accessed on 15 July 2021).
- Burbach, K.M.; Poland, A.; Bradfield, C.A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. USA 1992, 89, 8185–8189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poland, A.; Knutson, J.C. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 1982, 22, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Solomon, S.; Fraser, L.R.; Trombino, A.F.; Liu, D.; Sonenshein, G.E.; Hestermann, E.V.; Sherr, D.H. Constitutive regulation of CYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J. Cell Biochem. 2008, 104, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Yanagisawa, K.; Katafuchi, J.; Ringer, D.P.; Takami, Y.; Nakayama, T.; Suiko, M.; Liu, M.C. Molecular cloning, expression, and characterization of novel human SULT1C sulfotransferases that catalyze the sulfonation of N-hydroxy-2-acetylaminofluorene. J. Biol. Chem. 1998, 273, 33929–33935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar]
- Abugessaisa, I.; Ramilowski, J.A.; Lizio, M.; Severin, J.; Hasegawa, A.; Harshbarger, J.; Kondo, A.; Noguchi, S.; Yip, C.W.; Ooi, J.L.C.; et al. FANTOM enters 20th year: Expansion of transcriptomic atlases and functional annotation of non-coding RNAs. Nucleic Acids Res. 2021, 49, D892–D898. [Google Scholar] [CrossRef]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef] [Green Version]
- Bakulski, K.M.; Dou, J.; Lin, N.; London, S.J.; Colacino, J.A. DNA methylation signature of smoking in lung cancer is enriched for exposure signatures in newborn and adult blood. Sci. Rep. 2019, 9, 4576. [Google Scholar] [CrossRef] [Green Version]
- de Vries, M.; van der Plaat, D.A.; Nedeljkovic, I.; Verkaik-Schakel, R.N.; Kooistra, W.; Amin, N.; van Dujin, C.M.; Brandsma, C.; van Diemen, C.C.; Vonk, J.M.; et al. From blood to lung tissue: Effect of cigarette smoke on DNA methylation and lung function. Respir. Res. 2018, 19, 212. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LACTB Forward | 5′-GTTGAGAACCGTGTACCATGT-3′ |
| LACTB Reverse | 5′-TTCCCACAATTTGGCAAGAGC-3′ |
| SULT1C2 Forward | 5′-CAGCCTGCAACTGTGGACAA-3′ |
| SULT1C2 Reverse | 5′-GATGGCGGTGTTGGATGATG-3′ |
| CYP1B1 Forward | 5′-CTGCACTCGAGTCTGCACAT-3′ |
| CYP1B1 Reverse | 5′-TATCACTGACATCTTCGGCG-3′ |
| SULT1C2 promoter Forward | 5′-aaaaaaactagtCATCCCAGTTCATCCTCCACAAA-3′ |
| SULT1C2 promoter Reverse | 5′-aaaaaatcatgaTTTGAATAAATGCATCTGTAAAGCCA-3′ |
| MethyLight SULT1C2 Forward | 5′-GGGTATGGTGGCGTACGTT-3′ |
| MethyLight SULT1C2 Reverse | 5′-AATCTTAACTCACTACAACCTCCG-3′ |
| MethyLight SULT1C2 Probe | 5′-/6FAM-CTCCCGAATTCAAACGATTCTCCTATCTCA-BHQ-3/-3 |
| MethyLight ALU Forward | 5′-AGGTCGAGGTCGGCGG-3′ |
| MethyLight ALU Reverse | 5′-CCACGCCCGACTAATTTTATATCTT-3′ |
| MethyLight ALU Probe | 5′-/6FAM-CAAACTAATCTCAAACTCCCGACCTCAAACGA-BHQ-1/-3′ |
| ChIP SULT1C2 Forward | 5′-CCGTCTCTACTAAAAATACGAA-3′ |
| ChIP SULT1C2 Reverse | 5′-AGCGATTCTCCTGTCTCAGCC-3′ |
| ChIP CYP1B1 Forward | 5′-ATATGACTGGACCGACTTTCC-3′ |
| ChIP CYP1B1 Reverse | 5′-GGCGAACTTTATCGGGTTGA-3′ |
| SULT1C2 Expression | Number of Patients | Estimate | p-Value |
|---|---|---|---|
| Sample type | |||
| Normal | 53 | 0.60538 | 0.0479 |
| Tumor | 429 | ||
| Gender | |||
| Male | 210 | 0.13455 | 0.4926 |
| Female | 272 | ||
| Age | By Year | ||
| 482 | 0.01027 | 0.3031 | |
| Smoking | vs. Never Smoker | ||
| Never Smoker | 71 | -- | -- |
| Former Smoker (≥15 years) | 129 | −0.05579 | 0.8595 |
| Former Smoker (<15 years) | 169 | −0.70430 | 0.0187 |
| Current Smoker | 113 | −1.03470 | 0.0016 |
| Race | vs. Caucasian | ||
| Caucasian | 422 | -- | -- |
| Black or African American | 52 | 0.39562 | 0.2068 |
| Asian | 7 | 1.63644 | 0.0414 |
| American Indian or Alaskan Native | 1 | −0.12833 | 0.9512 |
| cg13968390 Methylation | Number of Patients | Estimate | p-Value |
|---|---|---|---|
| Race | vs. Caucasian | ||
| Caucasian | 359 | -- | -- |
| Black or African American | 51 | 0.005096 | 0.8020 |
| Asian | 6 | −0.130799 | 0.0197 |
| American Indian or Alaskan Native | 0 | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, C.; Mullen, D.J.; Selamat, S.A.; Campan, M.; Offringa, I.A.; Marconett, C.N. The Sulfotransferase SULT1C2 Is Epigenetically Activated and Transcriptionally Induced by Tobacco Exposure and Is Associated with Patient Outcome in Lung Adenocarcinoma. Int. J. Environ. Res. Public Health 2022, 19, 416. https://doi.org/10.3390/ijerph19010416
Johnson C, Mullen DJ, Selamat SA, Campan M, Offringa IA, Marconett CN. The Sulfotransferase SULT1C2 Is Epigenetically Activated and Transcriptionally Induced by Tobacco Exposure and Is Associated with Patient Outcome in Lung Adenocarcinoma. International Journal of Environmental Research and Public Health. 2022; 19(1):416. https://doi.org/10.3390/ijerph19010416
Chicago/Turabian StyleJohnson, Candace, Daniel J. Mullen, Suhaida A. Selamat, Mihaela Campan, Ite A. Offringa, and Crystal N. Marconett. 2022. "The Sulfotransferase SULT1C2 Is Epigenetically Activated and Transcriptionally Induced by Tobacco Exposure and Is Associated with Patient Outcome in Lung Adenocarcinoma" International Journal of Environmental Research and Public Health 19, no. 1: 416. https://doi.org/10.3390/ijerph19010416