
Removal of Ammonia Using Persulfate during the Nitrate Electro-Reduction Process

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Setup
2.2. Preparation of Particle Electrodes
2.3. Experimental Procedure
2.4. Identification of Reactive Species and Dominant Radicals
2.5. Analytical Methods
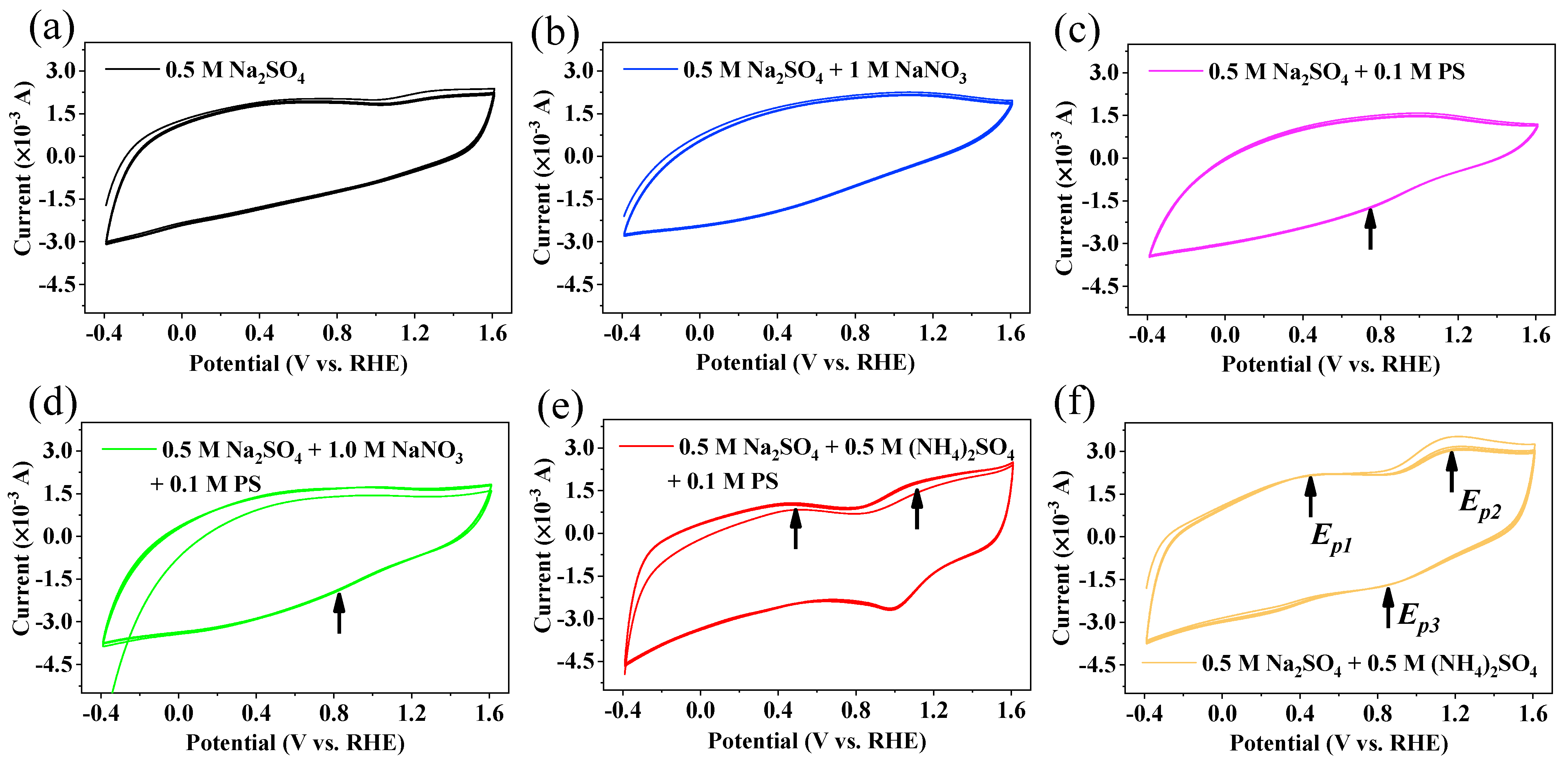
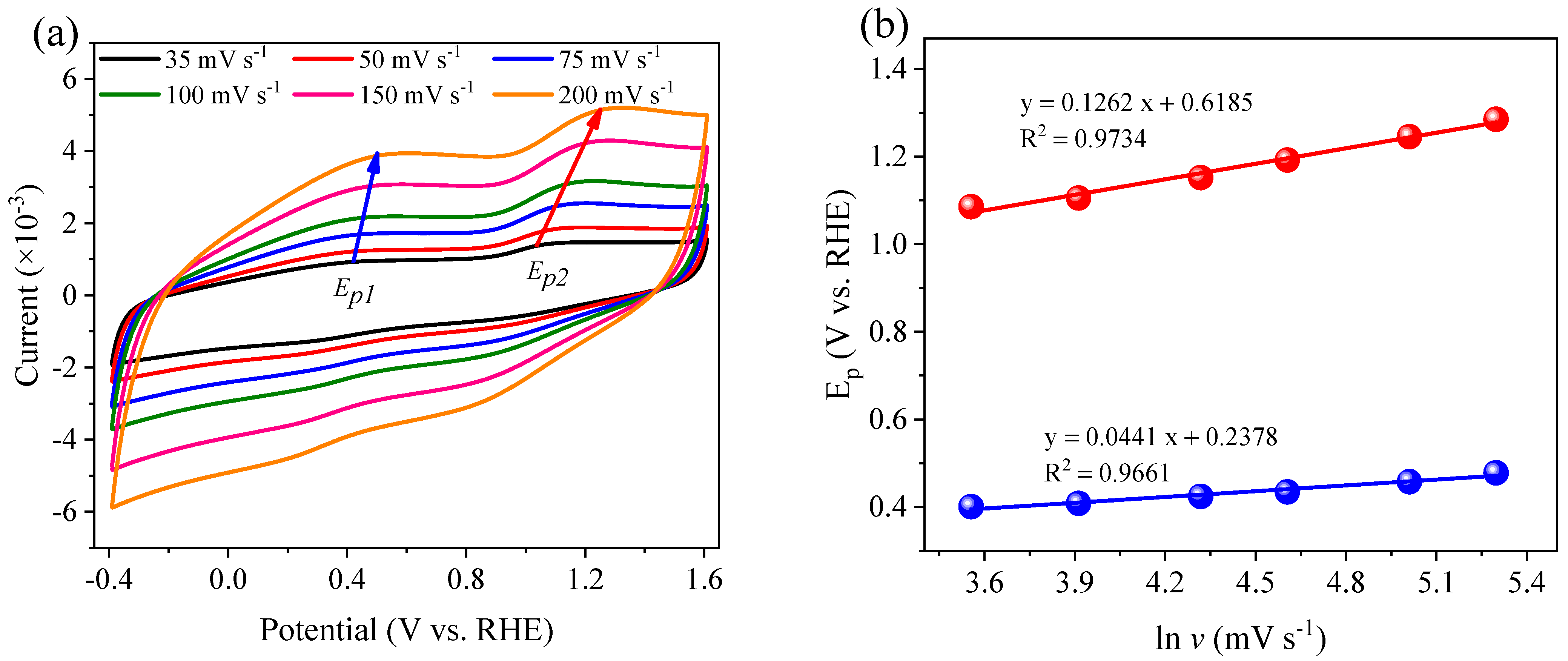
2.6. Electrochemical Measurements
2.7. Statistical Analysis
3. Results and Discussion
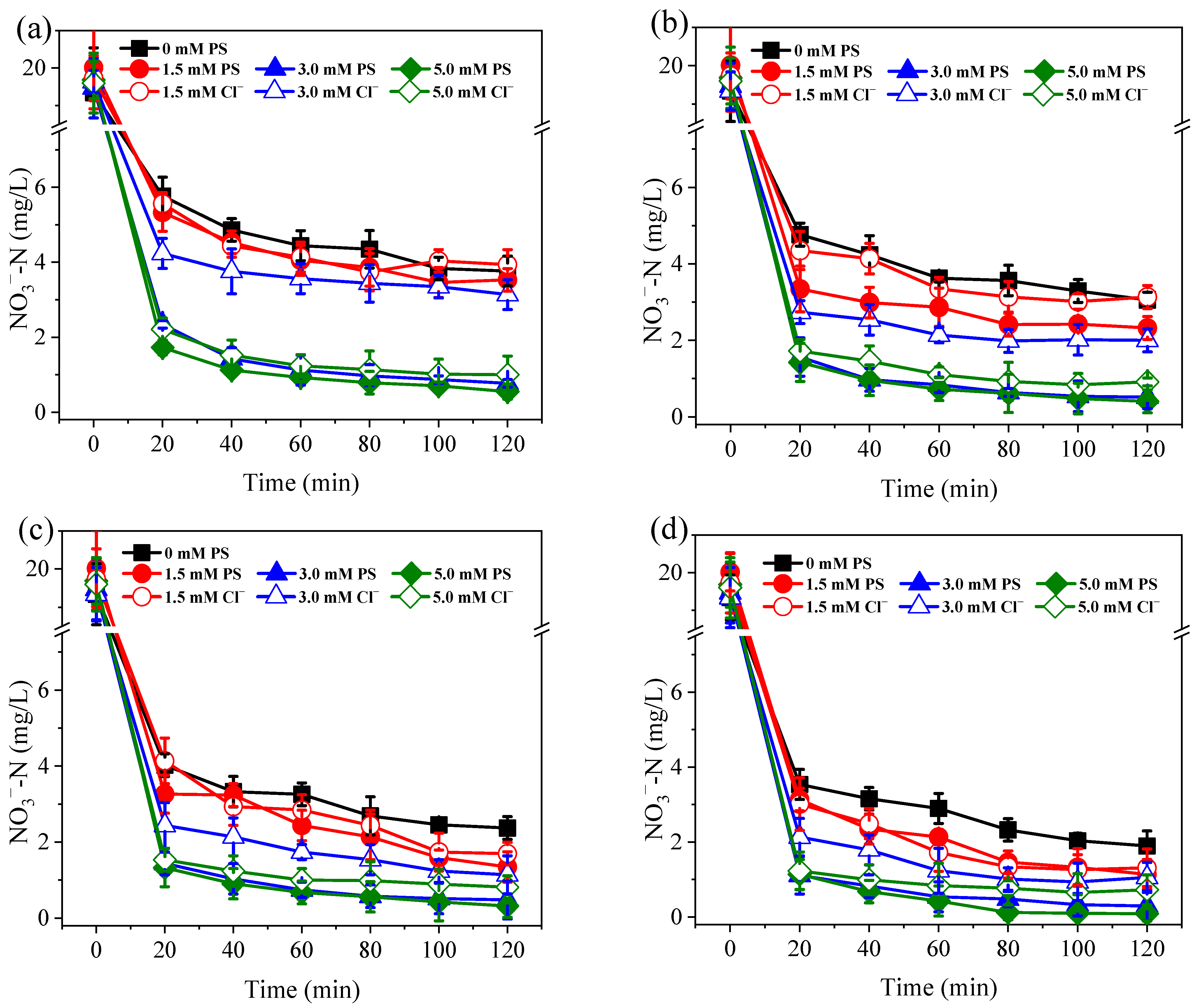
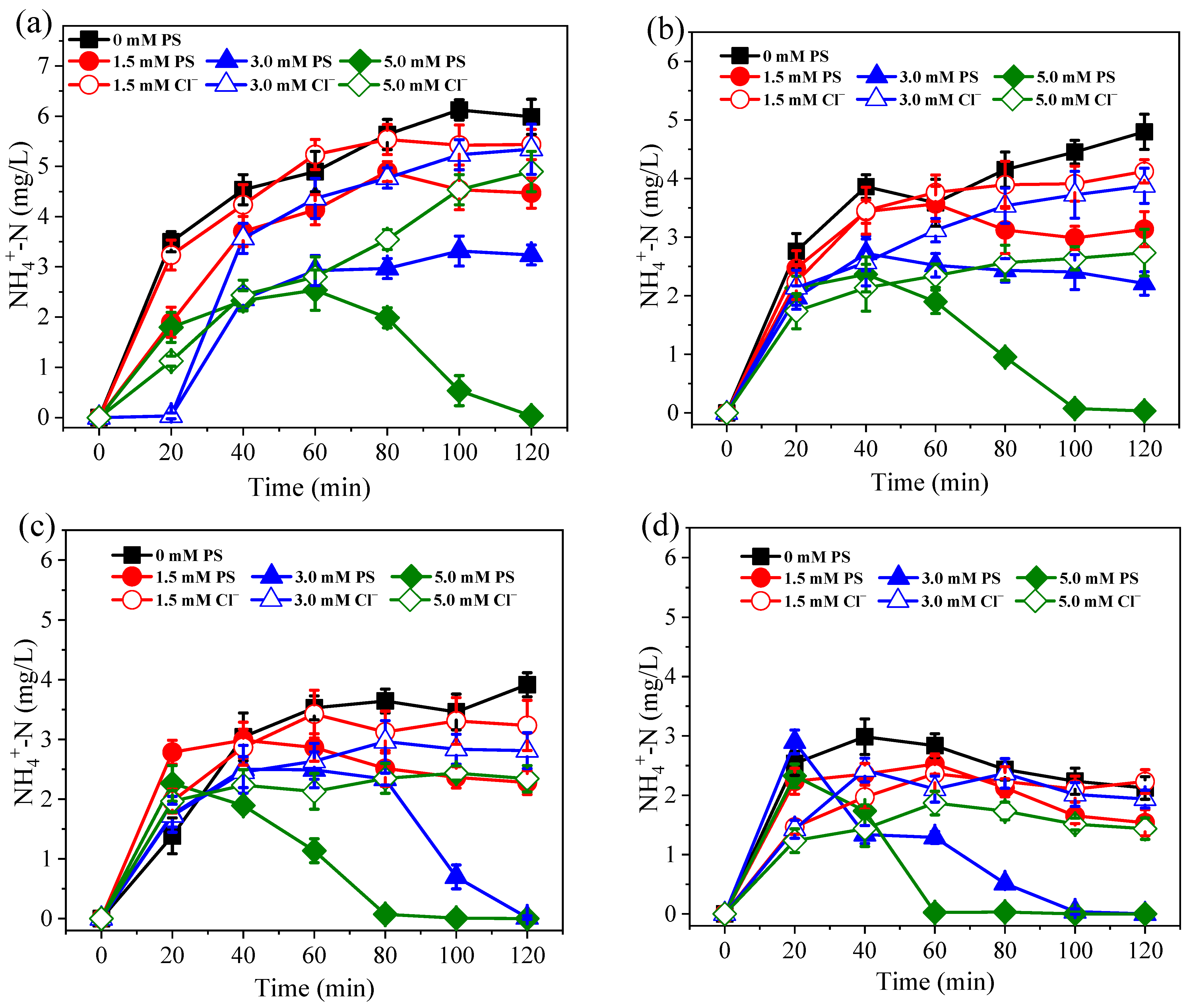
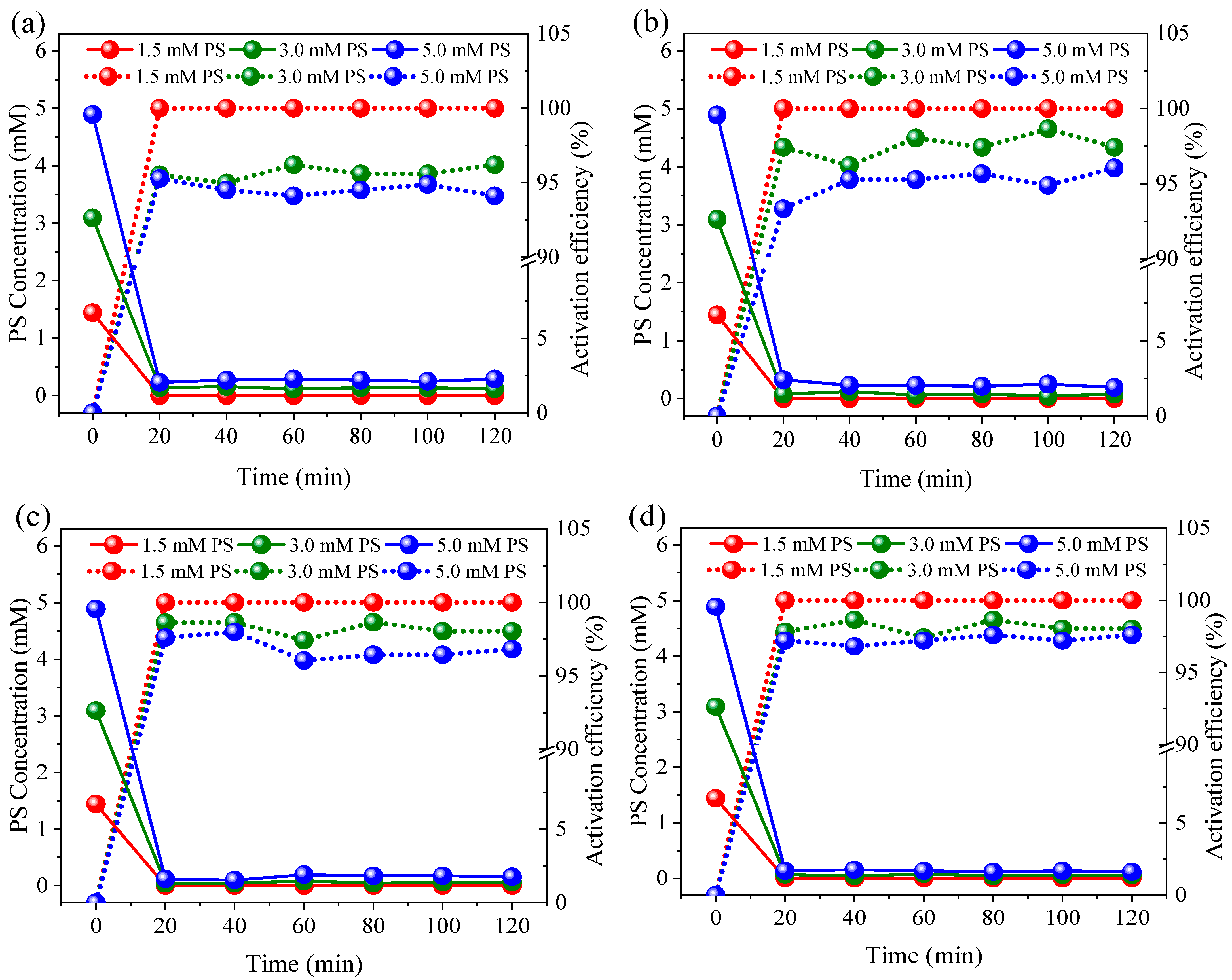
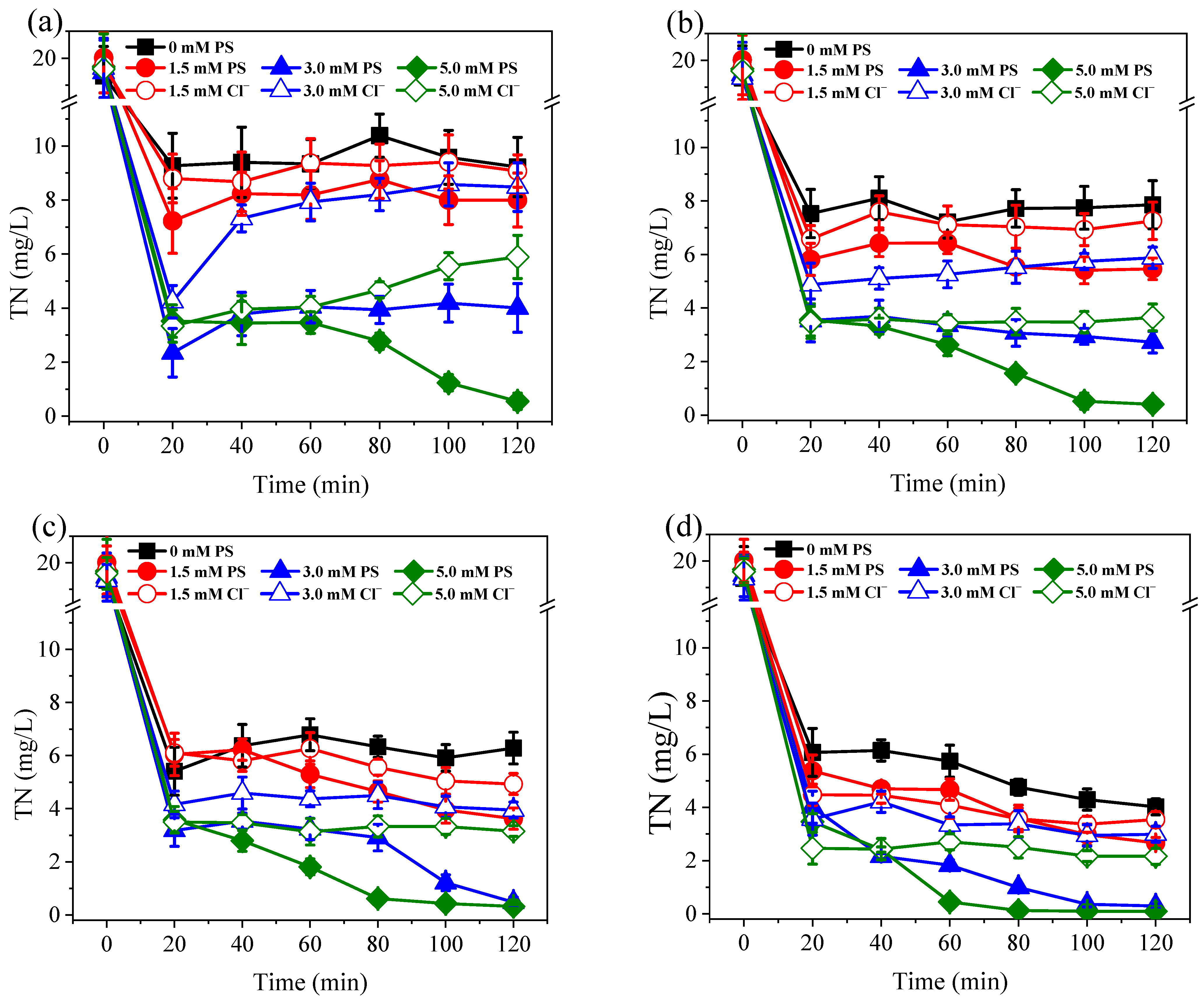
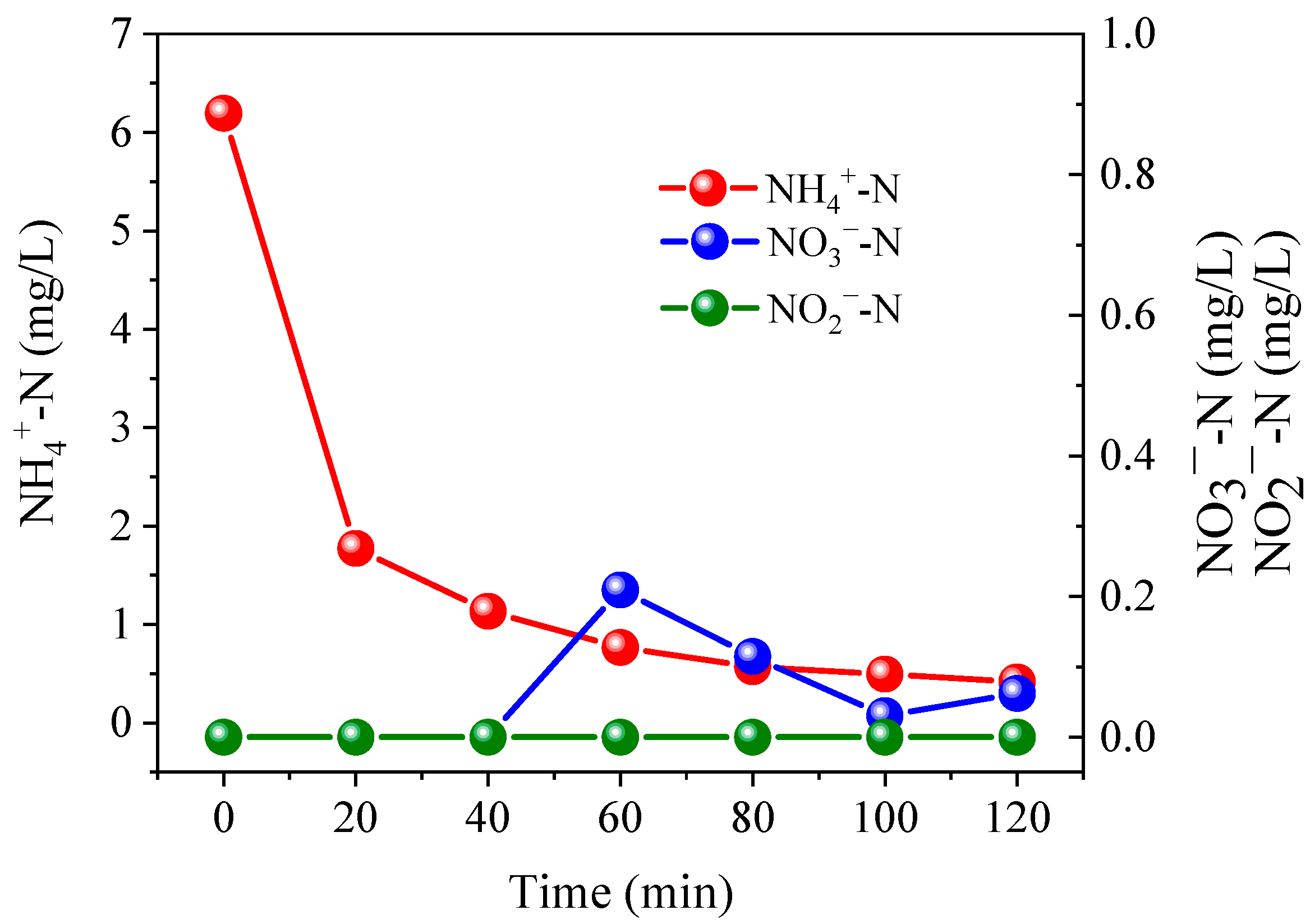
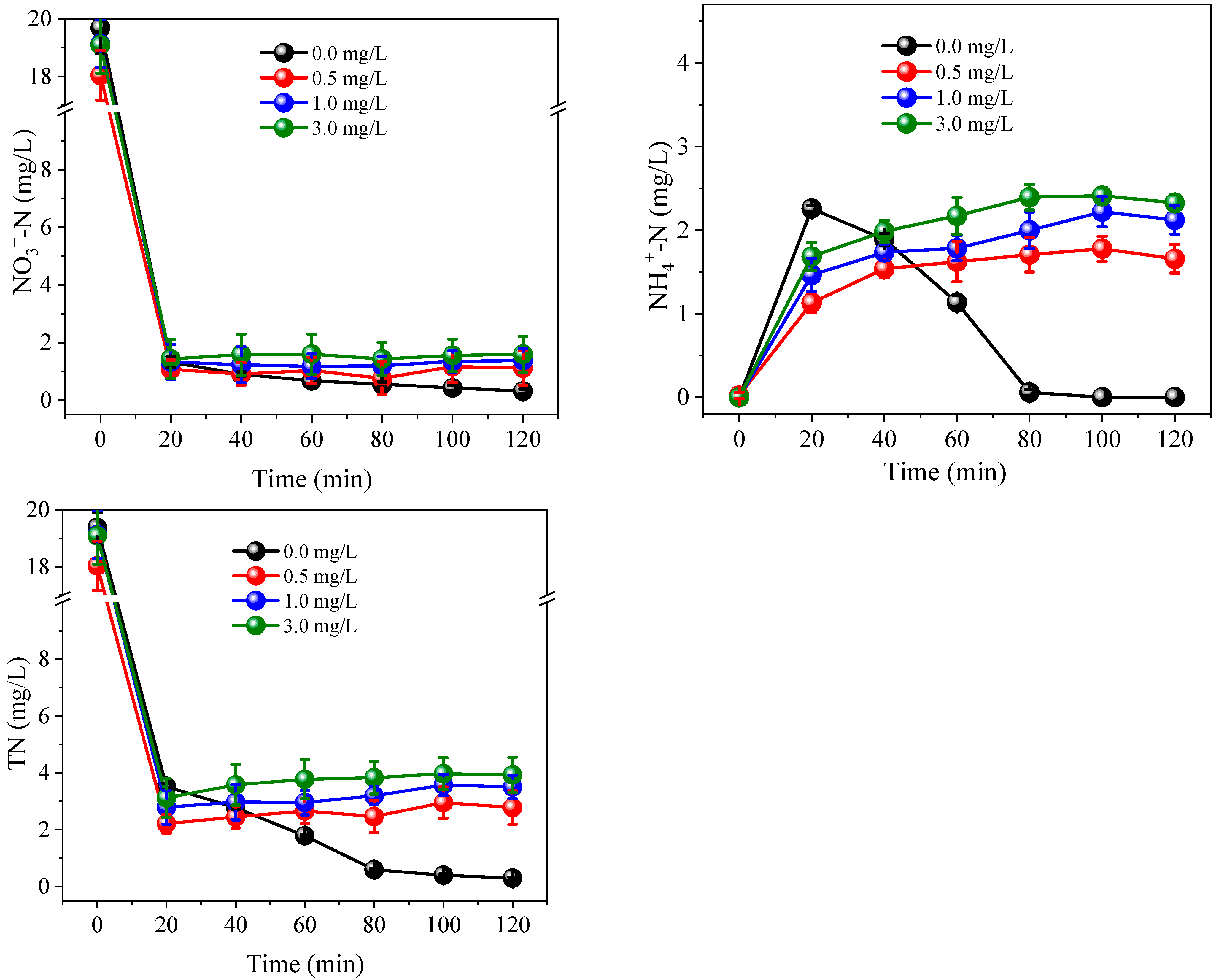
3.1. Nitrogen Conversion and Removal under Different PS Dosage and Current Conditions
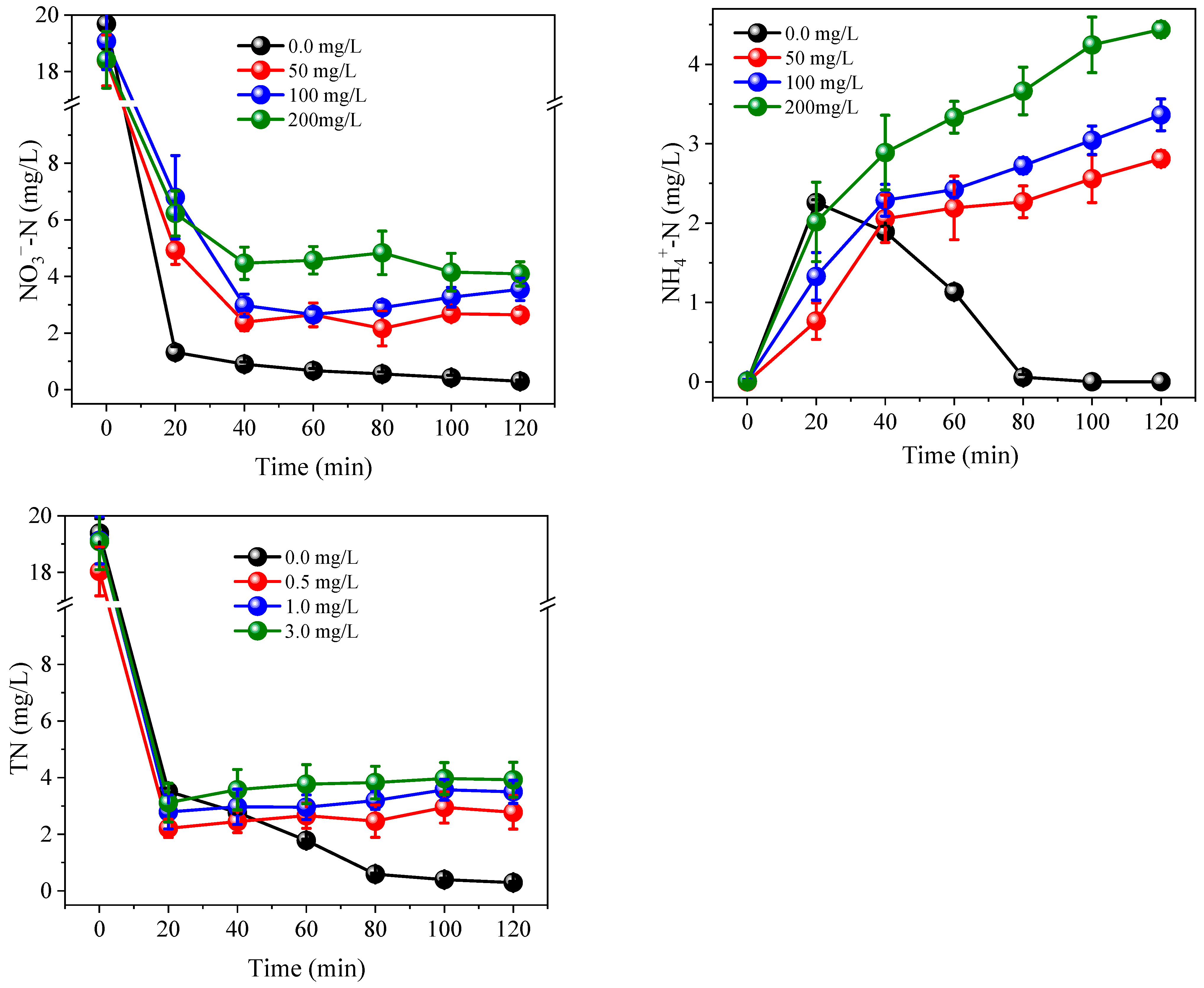
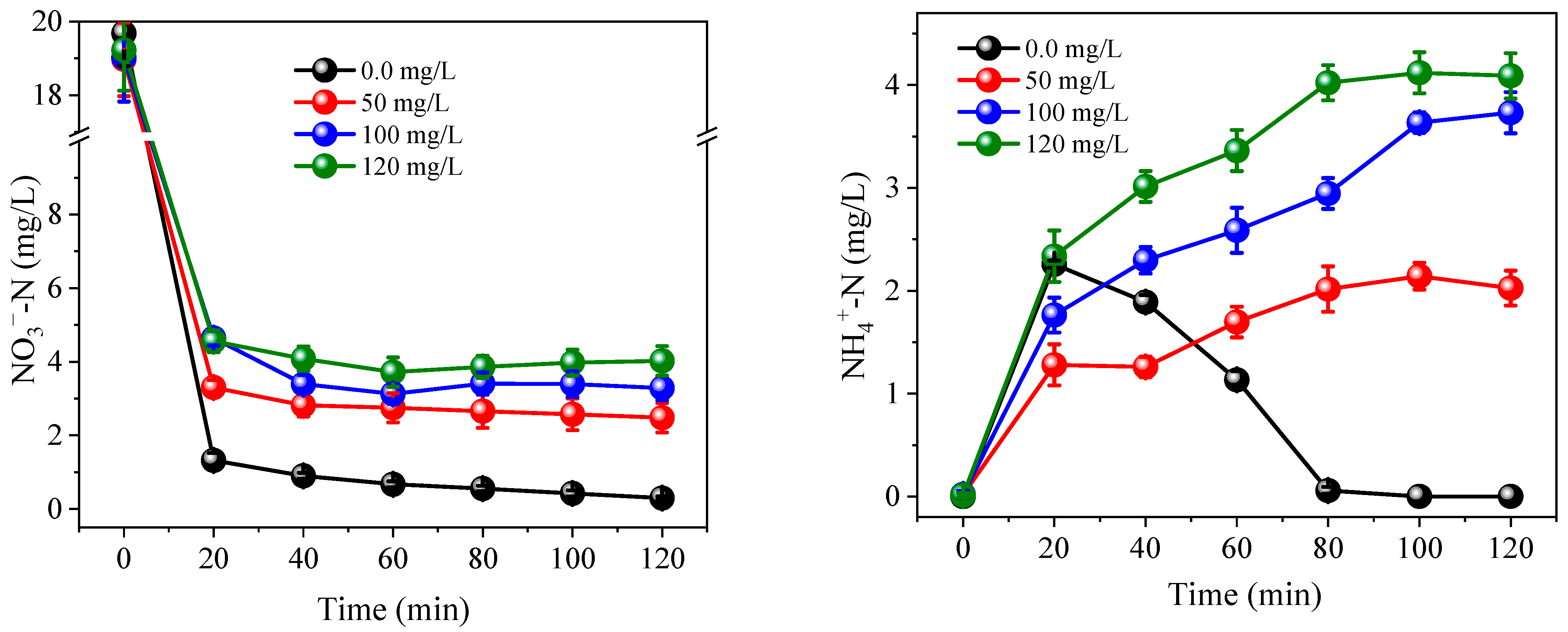
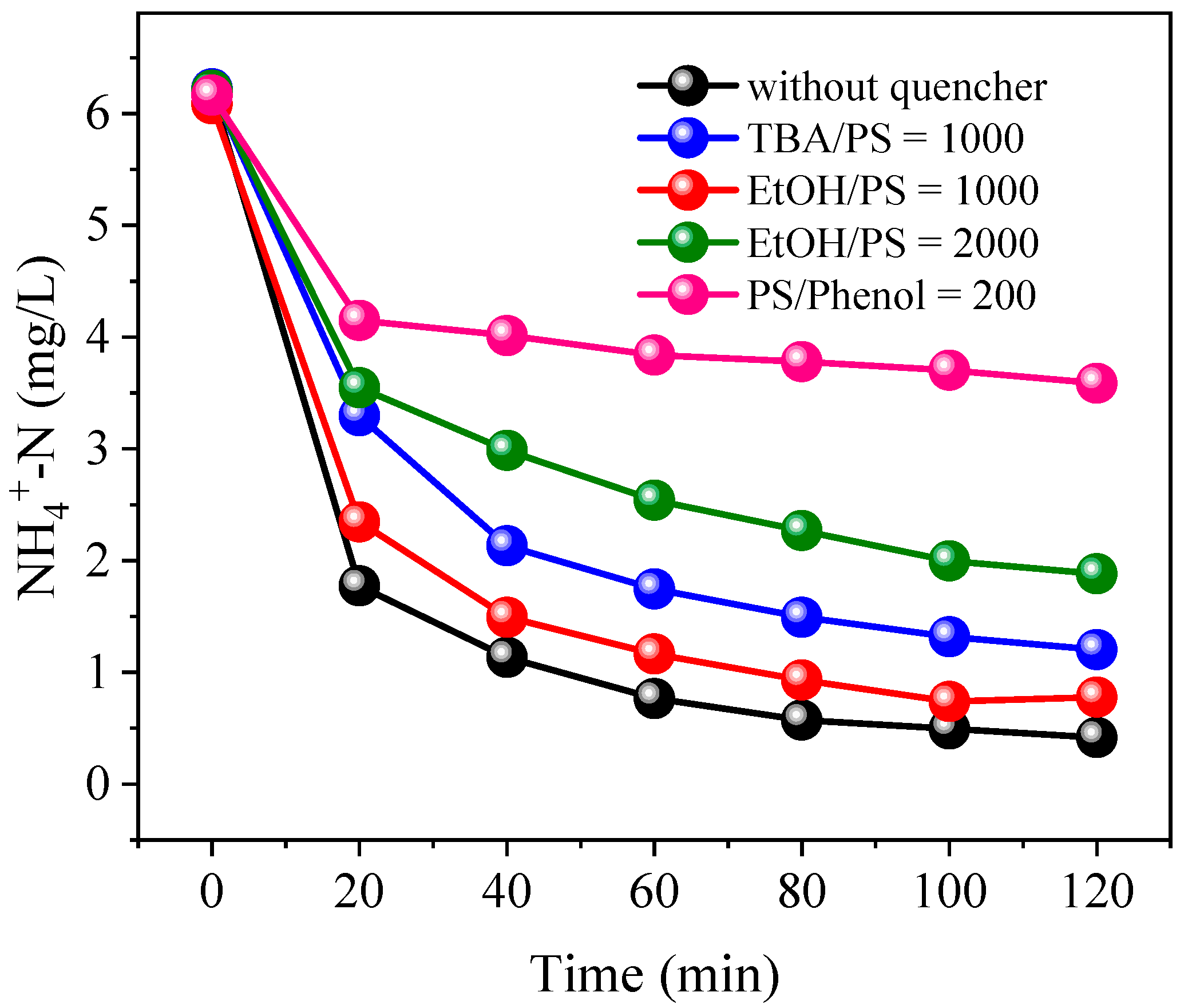
3.2. Effects of Coexisting Substances
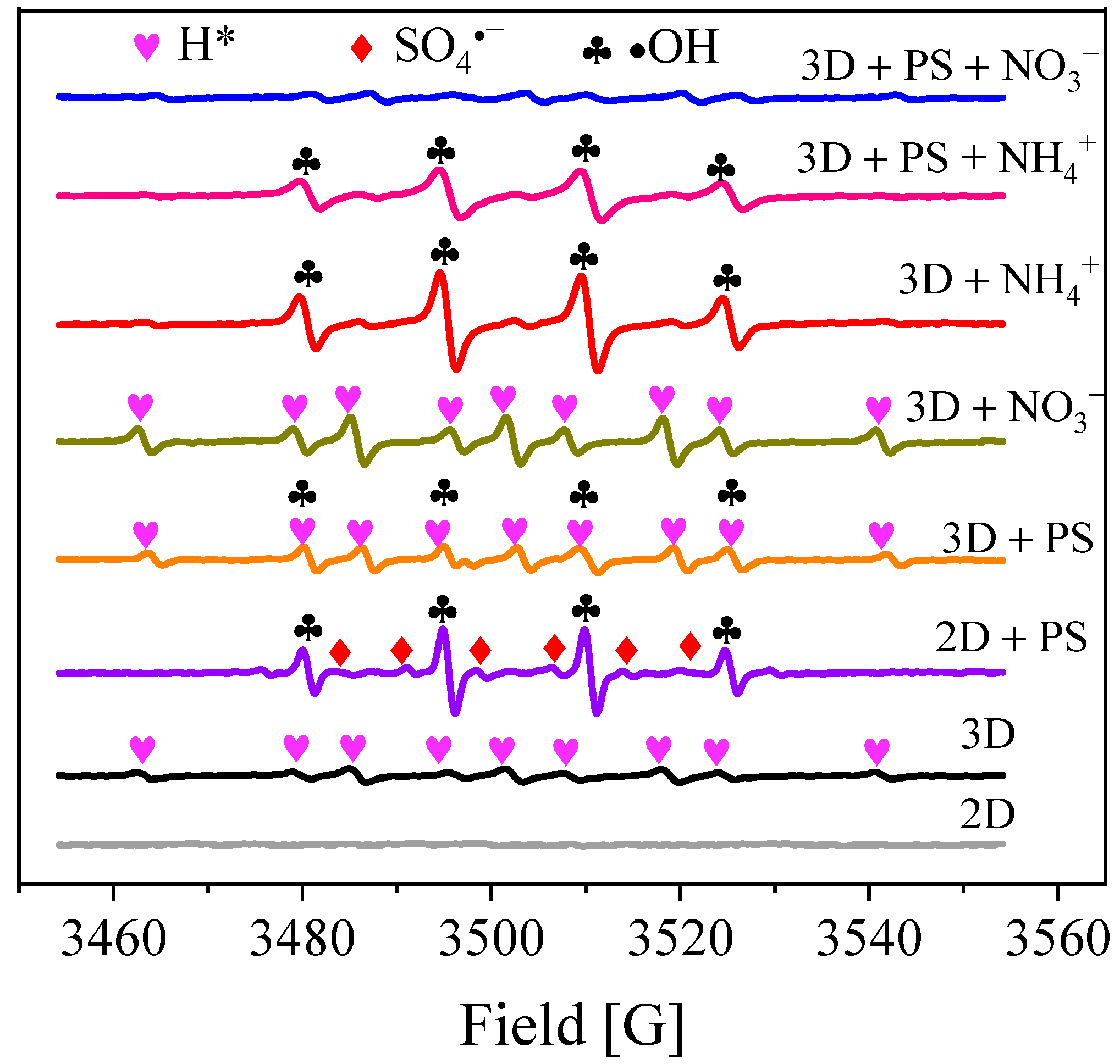
3.3. Reactive Species Identification
3.4. Identification of Dominant Radical
3.5. Electrocatalytic Performance of PS-Co/AC0.9-AB0.1
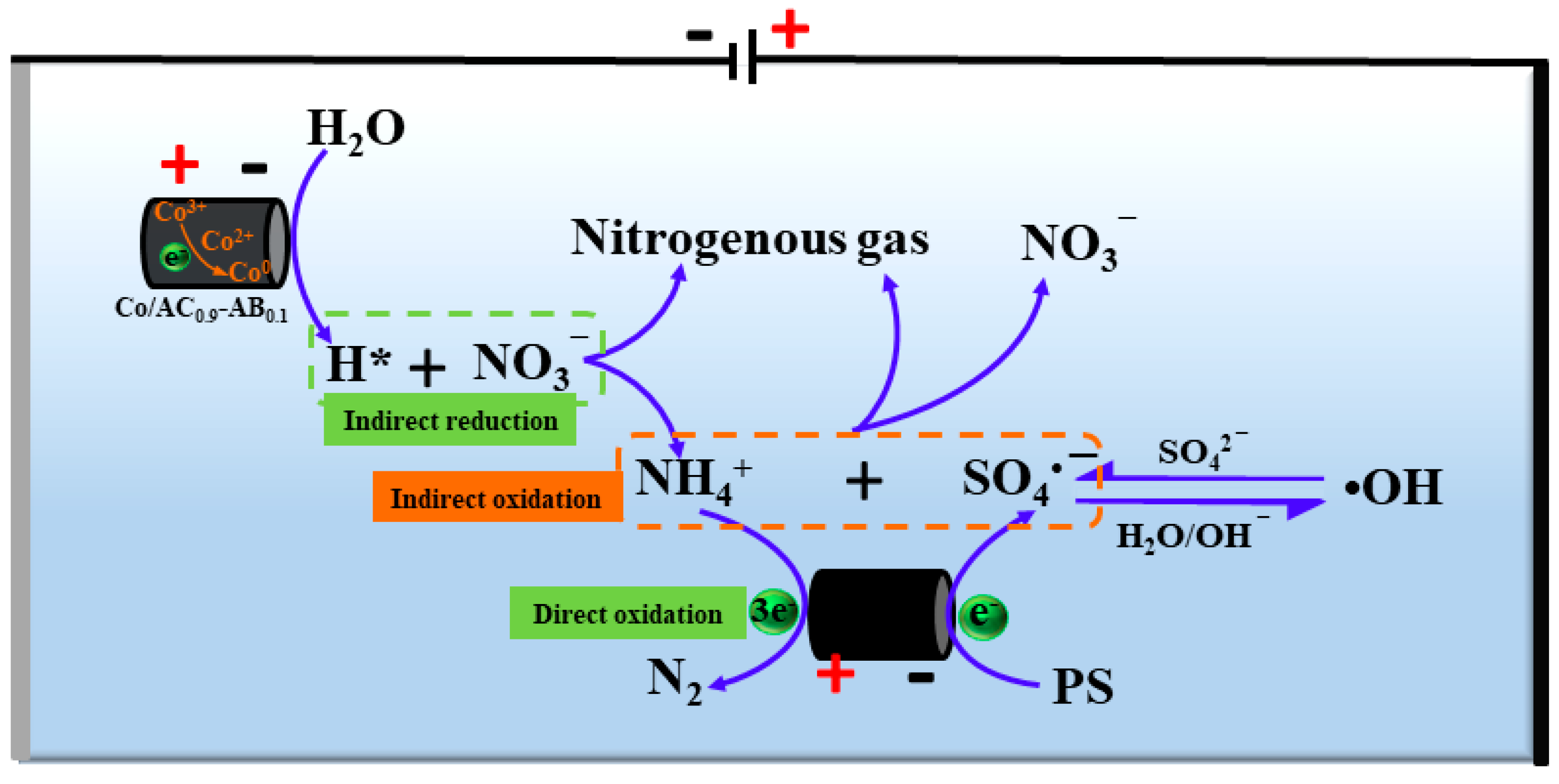
3.6. The Mechanism of N Transformation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, B.; Xu, X.; Ge, S.; Li, B.; Wei, Y.; Zhu, H.; Nan, X.; Peng, Y. Reducing carbon source consumption through a novel denitratation/anammox biofilter to remove nitrate from synthetic secondary effluent. Bioresour. Technol. 2020, 309, 123377. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Du, R.; Peng, Y.; Li, B.; Wang, S. Novel two stage partial denitrification (PD)-Anammox process for tertiary nitrogen removal from low carbon/nitrogen (C/N) municipal sewage. Chem. Eng. J. 2019, 362, 107–115. [Google Scholar] [CrossRef]
- Mohajeri, P.; Smith, C.M.S.; Chau, H.W.; Lehto, N. ALLODUST augmented activated sludge single batch anaerobic reactor (AS-SBAnR) for high concentration nitrate removal from agricultural wastewater. Sci. Total Environ. 2021, 752, 141905. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Kuan, W.-F.; Liu, H.; Huang, C.P. Mode of electrochemical deposition on the structure and morphology of bimetallic electrodes and its effect on nitrate reduction toward nitrogen selectivity. Appl. Catal. B Environ. 2019, 257, 117909. [Google Scholar] [CrossRef]
- Chauhan, R.; Srivastava, V.C. Electrochemical denitrification of highly contaminated actual nitrate wastewater by Ti/RuO2 anode and iron cathode. Chem. Eng. J. 2020, 386, 122065. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J. Reduction of nitrate by zero valent iron (ZVI)-based materials: A review. Sci. Total Environ. 2019, 671, 388–403. [Google Scholar] [CrossRef]
- Garcia-Segura, S.; Lanzarini-Lopes, M.; Hristovski, K.; Westerhoff, P. Electrocatalytic reduction of nitrate: Fundamentals to full-scale water treatment applications. Appl. Catal. B Environ. 2018, 236, 546–568. [Google Scholar] [CrossRef]
- Gao, J.; Jiang, B.; Ni, C.; Qi, Y.; Bi, X. Enhanced reduction of nitrate by noble metal-free electrocatalysis on P doped three-dimensional Co3O4 cathode: Mechanism exploration from both experimental and DFT studies. Chem. Eng. J. 2020, 382, 123034. [Google Scholar] [CrossRef]
- Gao, J.; Jiang, B.; Ni, C.; Yuanfeng, Q.; Zhang, Y.; Oturan, N.; Oturan, M. Non-precious Co3O4-TiO2/Ti cathode based electrocatalytic nitrate reduction: Preparation, performance and mechanism. Appl. Catal. B Environ. 2019, 254, 391–402. [Google Scholar] [CrossRef]
- Jin, J.; Wang, Y.; Wu, Z.; Hergazy, A.; Lan, J.; Zhao, L.; Liu, X.; Chen, N.; Lin, L. Transcriptomic analysis of liver from grass carp (Ctenopharyngodon idellus) exposed to high environmental ammonia reveals the activation of antioxidant and apoptosis pathways. Fish Shellfish Immunol. 2017, 63, 444–451. [Google Scholar] [CrossRef]
- Martínez, J.; Ortiz, A.; Ortiz, I. State-of-the-art and perspectives of the catalytic and electrocatalytic reduction of aqueous nitrates. Appl. Catal. B Environ. 2017, 207. [Google Scholar] [CrossRef] [Green Version]
- Dima, G.E.; de Vooys, A.C.A.; Koper, M.T.M. Electrocatalytic reduction of nitrate at low concentration on coinage and transition-metal electrodes in acid solutions. J. Electroanal. Chem. 2003, 554–555, 15–23. [Google Scholar] [CrossRef]
- Dash, B.P.; Chaudhari, S. Electrochemical denitrificaton of simulated ground water. Water Res. 2005, 39, 4065–4072. [Google Scholar] [CrossRef]
- Reyter, D.; Belanger, D.; Roue, L. Nitrate removal by a paired electrolysis on copper and Ti/IrO2 coupled electrodes—Influence of the anode/cathode surface area ratio. Water Res. 2010, 44, 1918–1926. [Google Scholar] [CrossRef]
- Mácová, Z.; Bouzek, K.; Šerák, J. Electrocatalytic activity of copper alloys for NO3− reduction in a weakly alkaline solution. J. Appl. Electrochem. 2007, 37, 557–566. [Google Scholar] [CrossRef]
- Reyter, D.; Bélanger, D.; Roué, L. Elaboration of Cu–Pd Films by Coelectrodeposition: Application to Nitrate Electroreduction. J. Phys. Chem. C 2008, 113, 290–297. [Google Scholar] [CrossRef]
- Birdja, Y.Y.; Yang, J.; Koper, M.T.M. Electrocatalytic Reduction of Nitrate on Tin-modified Palladium Electrodes. Electrochim. Acta 2014, 140, 518–524. [Google Scholar] [CrossRef]
- Comisso, N.; Cattarin, S.; Guerriero, P.; Mattarozzi, L.; Musiani, M.; Vázquez-Gómez, L.; Verlato, E. Study of Cu, Cu-Ni and Rh-modified Cu porous layers as electrode materials for the electroanalysis of nitrate and nitrite ions. J. Solid State Electrochem. 2015, 20, 1139–1148. [Google Scholar] [CrossRef]
- Lan, H.; Liu, X.; Liu, H.; Liu, R.; Hu, C.; Qu, J. Efficient Nitrate Reduction in a Fluidized Electrochemical Reactor Promoted by Pd–Sn/AC Particles. Catal. Lett. 2015, 146, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Xu, H.; Lv, M.; Wang, J.; Teng, W.; Ran, X.; Gou, X.; Wang, X.; Sun, Y.; Yang, J. Mesoporous carbon confined palladium–copper alloy composites for high performance nitrogen selective nitrate reduction electrocatalysis. New J. Chem. 2017, 41, 2349–2357. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, H.; Wang, L.; Chen, Y. Electrochemical removal of nitrate by Cu/Ti electrode coupled with copper-modified activated carbon particles at a low current density. Environ. Sci. Pollut. Res. Int. 2019, 26, 17567–17576. [Google Scholar] [CrossRef]
- Ye, W.; Zhang, W.; Hu, X.; Yang, S.; Liang, W. Efficient electrochemical-catalytic reduction of nitrate using Co/AC0.9-AB0.1 particle electrode. Sci. Total Environ. 2020, 732, 139245. [Google Scholar] [CrossRef]
- Sun, W.; Sun, Y.; Shah, K.J.; Chiang, P.C.; Zheng, H. Electrocatalytic oxidation of tetracycline by Bi-Sn-Sbγ-Al2O3 three-dimensional particle electrode. J. Hazard. Mater. 2019, 370, 24–32. [Google Scholar] [CrossRef]
- Mandal, P.; Yadav, M.K.; Gupta, A.K.; Dubey, B.K. Chlorine mediated indirect electro-oxidation of ammonia using non-active PbO2 anode: Influencing parameters and mechanism identification. Sep. Purif. Technol. 2020, 247, 116910. [Google Scholar] [CrossRef]
- Devi, P.; Dalai, A.K. Implications of breakpoint chlorination on chloramines decay and disinfection by-products formation in brine solution. Desalination 2021, 504, 114961. [Google Scholar] [CrossRef]
- Huang, L.; Li, L.; Dong, W.; Liu, Y.; Hou, H. Removal of Ammonia by OH Radical in Aqueous Phase. Environ. Sci.Technol. 2008, 42, 8070–8075. [Google Scholar] [CrossRef]
- Wacławek, S.; Lutze, H.V.; Grübel, K.; Padil, V.V.T.; Černík, M.; Dionysiou, D.D. Chemistry of persulfates in water and wastewater treatment: A review. Chem. Eng. J. 2017, 330, 44–62. [Google Scholar] [CrossRef]
- Van Berkel, K.Y.; Russell, G.T.; Gilbert, R.G. The dissociation rate coefficient of persulfate in emulsion polymerization systems. Polymer 2006, 47, 4667–4675. [Google Scholar] [CrossRef]
- Du, X.; Zhou, M. Strategies to enhance catalytic performance of metal-organic frameworks in sulfate radical-based advanced oxidation processes for organic pollutants removal. Chem. Eng. J. 2021, 403, 126346. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, H. Mn-based catalysts for sulfate radical-based advanced oxidation processes: A review. Environ. Int. 2019, 133, 105141. [Google Scholar] [CrossRef]
- Giannakis, S.; Lin, K.-Y.A.; Ghanbari, F. A review of the recent advances on the treatment of industrial wastewaters by Sulfate Radical-based Advanced Oxidation Processes (SR-AOPs). Chem. Eng. J. 2021, 406, 127083. [Google Scholar] [CrossRef]
- Chen, C.; Feng, H.; Deng, Y. Re-evaluation of sulfate radical based-advanced oxidation processes (SR-AOPs) for treatment of raw municipal landfill leachate. Water Res. 2019, 153, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sun, Z.; Cui, J. Research on the mechanism and reaction conditions of electrochemical preparation of persulfate in a split-cell reactor using BDD anode. RSC Adv. 2020, 10, 33928–33936. [Google Scholar] [CrossRef]
- Qing, G.; Ghazfar, R.; Jackowski, S.T.; Habibzadeh, F.; Ashtiani, M.M.; Chen, C.P.; Smith, M.R., 3rd; Hamann, T.W. Recent Advances and Challenges of Electrocatalytic N2 Reduction to Ammonia. Chem. Rev. 2020, 120, 5437–5516. [Google Scholar] [CrossRef]
- Liang, C.; Huang, C.-F.; Mohanty, N.; Kurakalva, R.M. A rapid spectrophotometric determination of persulfate anion in ISCO. Chemosphere 2008, 73, 1540–1543. [Google Scholar] [CrossRef]
- Zhang, W.; He, Y.; Li, C.; Hu, X.; Yang, S.; You, X.; Liang, W. Persulfate activation using Co/AC particle electrodes and synergistic effects on humic acid degradation. Appl. Catal. B Environ. 2021, 285, 119848. [Google Scholar] [CrossRef]
- Yan, Z.; Zheng, X.; Fan, J.; Zhang, Y.; Wang, S.; Zhang, T.; Sun, Q.; Huang, Y. China national water quality criteria for the protection of freshwater life: Ammonia. Chemosphere 2020, 251, 126379. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, S.; Chen, D.; Dai, G.; Wei, D.; Shu, Y. Activation of persulfate with biochar for degradation of bisphenol A in soil. Chem. Eng. J. 2020, 381, 122637. [Google Scholar] [CrossRef]
- Huang, W.; Xiao, S.; Zhong, H.; Yan, M.; Yang, X. Activation of persulfates by carbonaceous materials: A review. Chem. Eng. J. 2021, 418, 129297. [Google Scholar] [CrossRef]
- Wang, X.-H.; Wang, X.; Huppes, G.; Heijungs, R.; Ren, N.-Q. Environmental implications of increasingly stringent sewage discharge standards in municipal wastewater treatment plants: Case study of a cool area of China. J. Clean. Prod. 2015, 94, 278–283. [Google Scholar] [CrossRef]
- Chen, L.; Hu, X.; Cai, T.; Yang, Y.; Zhao, R.; Liu, C.; Li, A.; Jiang, C. Degradation of Triclosan in soils by thermally activated persulfate under conditions representative of in situ chemical oxidation (ISCO). Chem. Eng. J. 2019, 369, 344–352. [Google Scholar] [CrossRef]
- Qi, F.; Zeng, Z.; Wen, Q.; Huang, Z. Enhanced organics degradation by three-dimensional (3D) electrochemical activation of persulfate using sulfur-doped carbon particle electrode: The role of thiophene sulfur functional group and specific capacitance. J. Hazard. Mater. 2021, 416, 125810. [Google Scholar] [CrossRef]
- Zhu, X.; Nanny, M.; Butler, E. Effects of pH and Catalyst Concentration on Photocatalytic Oxidation of Aqueous Ammonia and Nitrite in Titanium Dioxide Suspensions. Environ.Sci.Technol. 2005, 39, 3784–3791. [Google Scholar] [CrossRef]
- Gong, C.; Chen, F.; Yang, Q.; Luo, K.; Yao, F.; Wang, S.; Wang, X.; Wu, J.; Li, X.; Wang, D.; et al. Heterogeneous activation of peroxymonosulfate by Fe-Co layered doubled hydroxide for efficient catalytic degradation of Rhoadmine B. Chem. Eng. J. 2017, 321, 222–232. [Google Scholar] [CrossRef]
- Yang, S.; Yang, X.; Shao, X.; Niu, R.; Wang, L. Activated carbon catalyzed persulfate oxidation of Azo dye acid orange 7 at ambient temperature. J. Hazard. Mater. 2011, 186, 659–666. [Google Scholar] [CrossRef]
- Xu, Y.; Lin, H.; Li, Y.; Zhang, H. The mechanism and efficiency of MnO2 activated persulfate process coupled with electrolysis. Sci. Total Environ. 2017, 609, 644–654. [Google Scholar] [CrossRef]
- Yan, S.; Xiong, W.; Xing, S.; Shao, Y.; Guo, R.; Zhang, H. Oxidation of organic contaminant in a self-driven electro/natural maghemite/peroxydisulfate system: Efficiency and mechanism. Sci. Total Environ. 2017, 599–600, 1181–1190. [Google Scholar] [CrossRef]
- Oh, H.K.; Takizawa, S.; Ohgaki, S.; Katayama, H.; Oguma, K.; Yu, M.J. Removal of organics and viruses using hybrid ceramic MF system without draining PAC. Desalination 2007, 202, 191–198. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, X.; Yang, Y.; Feng, Y.; Wu, D.; Mao, S. Activation of persulfate with metal–organic framework-derived nitrogen-doped porous Co@C nanoboxes for highly efficient p-Chloroaniline removal. Chem. Eng. J. 2019, 358, 408–418. [Google Scholar] [CrossRef]
- Qian, L.; Gu, L.; Yuan, H.; Xiao, D. Direct growth of NiCo2O4 nanostructures on conductive substrates with enhanced electrocatalytic activity and stability for methanol oxidation. Nanoscale 2013, 5, 7388–7396. [Google Scholar] [CrossRef]
- Wang, H.; Qing, C.; Guo, J.; Aref, A.A.; Sun, D.; Wang, B.; Tang, Y. Highly conductive carbon–CoO hybrid nanostructure arrays with enhanced electrochemical performance for asymmetric supercapacitors. J. Mater. Chem. A 2014, 2, 11776–11783. [Google Scholar] [CrossRef]
- Li, A.; Zhao, X.; Hou, Y.; Liu, H.; Wu, L.; Qu, J. The electrocatalytic dechlorination of chloroacetic acids at electrodeposited Pd/Fe-modified carbon paper electrode. Appl. Catal. B Environ. 2012, 111–112, 628–635. [Google Scholar] [CrossRef]
- Duan, X.; Su, C.; Zhou, L.; Sun, H.; Suvorova, A.; Odedairo, T.; Zhu, Z.; Shao, Z.; Wang, S. Surface controlled generation of reactive radicals from persulfate by carbocatalysis on nanodiamonds. Appl. Catal. B Environ. 2016, 194, 7–15. [Google Scholar] [CrossRef]
- Hu, X.; Yang, S.; You, X.; Zhang, W.; Liu, Y.; Liang, W. Electrocatalytic Decomplexation of Cu-EDTA and Simultaneous Recovery of Cu with Ni/GO-PAC Particle Electrode. Chem. Eng. J. 2021, 428, 131468. [Google Scholar] [CrossRef]
- Laviron, E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 1979, 101, 19–28. [Google Scholar] [CrossRef]
- Luong, J.H.T.; Glennon, J.D.; Gedanken, A.; Vashist, S.K. Achievement and assessment of direct electron transfer of glucose oxidase in electrochemical biosensing using carbon nanotubes, graphene, and their nanocomposites. Microchim. Acta 2016, 184, 369–388. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Hu, X.; You, X.; Zhang, W.; Liu, Y.; Liang, W. Removal of Ammonia Using Persulfate during the Nitrate Electro-Reduction Process. Int. J. Environ. Res. Public Health 2022, 19, 3270. https://doi.org/10.3390/ijerph19063270
Yang S, Hu X, You X, Zhang W, Liu Y, Liang W. Removal of Ammonia Using Persulfate during the Nitrate Electro-Reduction Process. International Journal of Environmental Research and Public Health. 2022; 19(6):3270. https://doi.org/10.3390/ijerph19063270
Chicago/Turabian StyleYang, Shuai, Xinxin Hu, Xinyu You, Wenwen Zhang, Yu Liu, and Wenyan Liang. 2022. "Removal of Ammonia Using Persulfate during the Nitrate Electro-Reduction Process" International Journal of Environmental Research and Public Health 19, no. 6: 3270. https://doi.org/10.3390/ijerph19063270