
Conversion of Polyethylene to High-Yield Fuel Oil at Low Temperatures and Atmospheric Initial Pressure
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
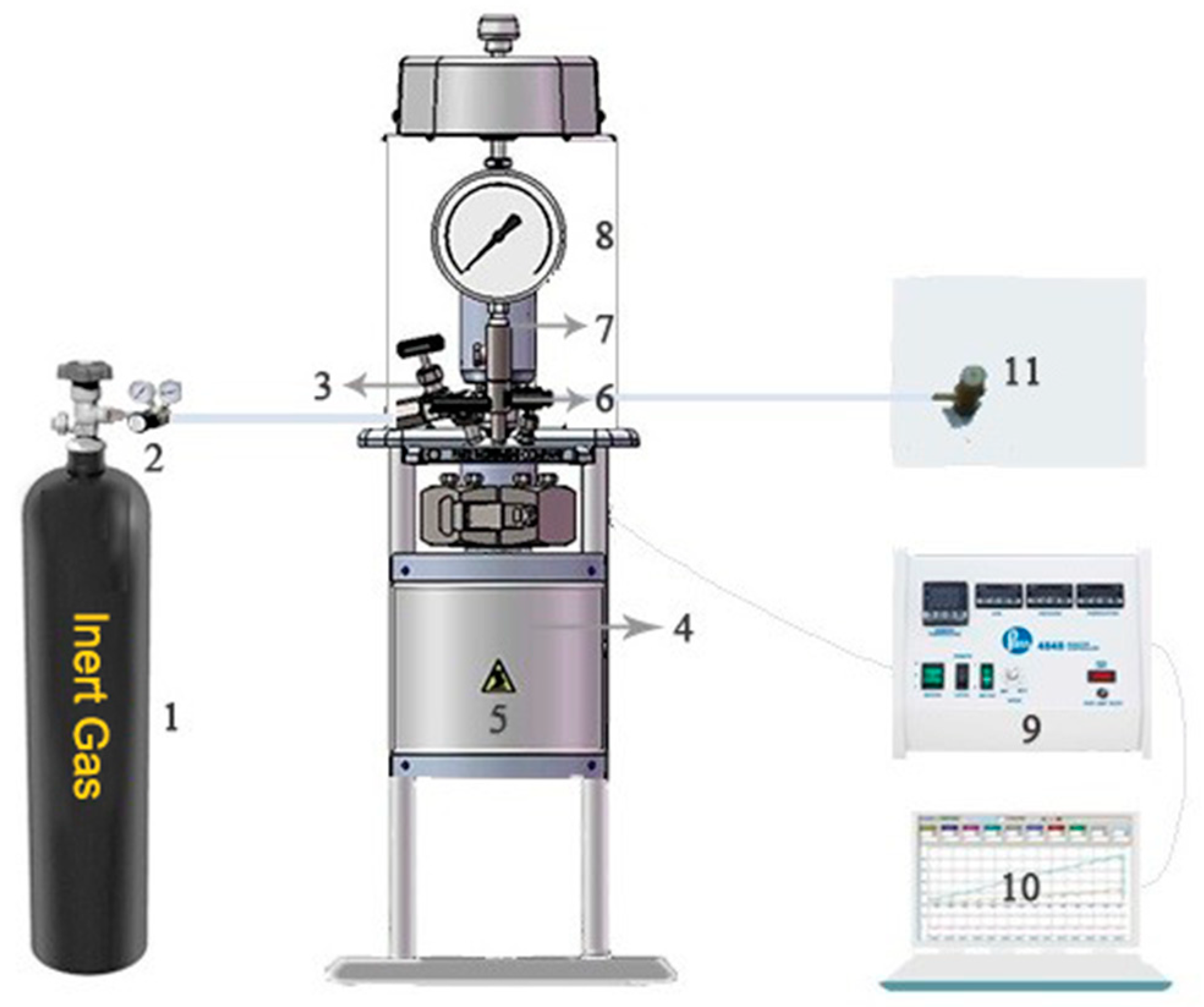
2.2. Experimental Setup and Operating Conditions
2.3. Analytical Methods
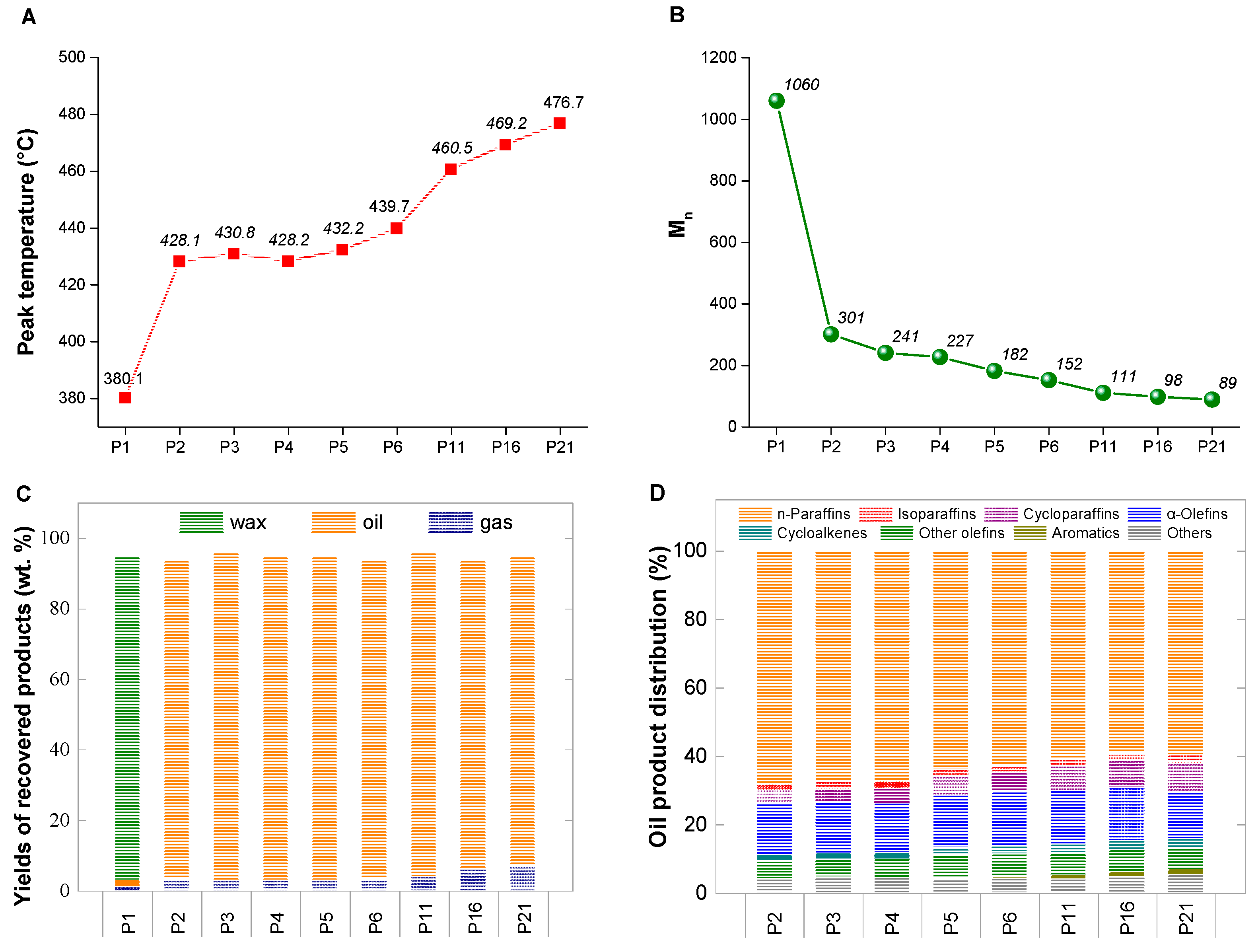
3. Results
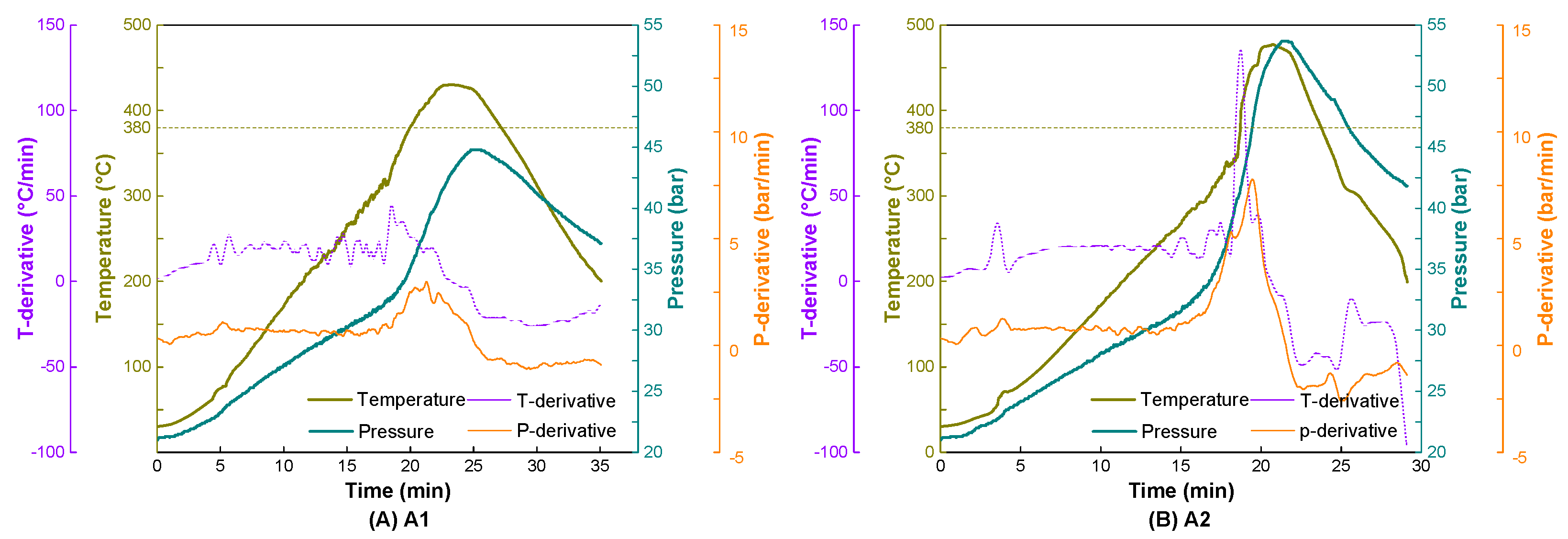
3.1. The Effect of Nitrogen Pressure
3.2. Effect of the Different Atmospheres
3.3. Promotion or Inhibition Effect of Low-Boiling Hydrocarbons on Pyrolysis
4. Environmental and Health Issues in Plastic Waste Management
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, A.A.; Dankwa Ampah, J.; Soudagar, M.E.M.; Veza, I.; Kingsley, U.; Afrane, S.; Jin, C.; Liu, H.; Elfasakhany, A.; Buyondo, K.A. Effects of hybrid nanoparticle additives in n-butanol/waste plastic oil/diesel blends on combustion, particulate and gaseous emissions from diesel engine evaluated with entropy-weighted PROMETHEE II and TOPSIS: Environmental and health risks of plastic waste. Energy Convers. Manag. 2022, 264, 115758. [Google Scholar]
- Weithmann, N.; Möller, J.N.; Löder, M.G.J.; Piehl, S.; Laforsch, C.; Freitag, R. Organic fertilizer as a vehicle for the entry of microplastic into the environment. Sci. Adv. 2018, 4, eaap8060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Li, B.; Zou, X.; Wang, Y.; Li, Y.; Xu, Y.; Mao, L.; Zhang, C.; Yu, W. Emission of primary microplastics in mainland China: Invisible but not negligible. Water Res. 2019, 162, 214–224. [Google Scholar] [CrossRef]
- Jiao, X.; Zheng, K.; Hu, Z.; Zhu, S.; Sun, Y.; Xie, Y. Conversion of Waste Plastics into Value-Added Carbonaceous Fuels under Mild Conditions. Adv. Mater. 2021, 33, 2005192. [Google Scholar] [CrossRef]
- Vollmer, I.; Jenks, M.J.F.; Roelands, M.C.P.; White, R.J.; van Harmelen, T.; de Wild, P.; van der Laan, G.P.; Meirer, F.; Keurentjes, J.T.F.; Weckhuysen, B.M. Beyond Mechanical Recycling: Giving New Life to Plastic Waste. Angew. Chem. Int. Ed. 2020, 59, 15402–15423. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Guo, J.; Zhang, W.; Summers, P.A.; Hall, P. From waste plastics to industrial raw materials: A life cycle assessment of mechanical plastic recycling practice based on a real-world case study. Sci. Total Environ. 2017, 601–602, 1192–1207. [Google Scholar] [CrossRef] [PubMed]
- Kratish, Y.; Li, J.; Liu, S.; Gao, Y.; Marks, T.J. Polyethylene Terephthalate Deconstruction Catalyzed by a Carbon-Supported Single-Site Molybdenum-Dioxo Complex. Angew. Chem. Int. Ed. 2020, 59, 19857–19861. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; von Wolff, N.; Rauch, M.; Zou, Y.-Q.; Shmul, G.; Ben-David, Y.; Leitus, G.; Avram, L.; Milstein, D. Hydrogenative Depolymerization of Nylons. J. Am. Chem. Soc. 2020, 142, 14267–14275. [Google Scholar] [CrossRef] [PubMed]
- Coates, G.W.; Getzler, Y. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater. 2020, 5, 501–516. [Google Scholar] [CrossRef]
- Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and chemical recycling of solid plastic waste. Waste Manag. 2017, 69, 24–58. [Google Scholar] [CrossRef]
- Zhou, C.; Fang, W.; Xu, W.; Cao, A.; Wang, R. Characteristics and the recovery potential of plastic wastes obtained from landfill mining. J. Clean. Prod. 2014, 80, 80–86. [Google Scholar] [CrossRef]
- Santos, E.; Rijo, B.; Lemos, F.; Lemos, M.A.N.D.A. A catalytic reactive distillation approach to high density polyethylene pyrolysis—Part 1-Light olefin production. Chem. Eng. J. 2019, 378, 122077. [Google Scholar] [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Navarro, R. Evolution of products during the degradation of polyethylene in a batch reactor. J. Anal. Appl. Pyrolysis 2009, 86, 14–21. [Google Scholar] [CrossRef]
- Onwudili, J.A.; Insura, N.; Williams, P.T. Composition of products from the pyrolysis of polyethylene and polystyrene in a closed batch reactor: Effects of temperature and residence time. J. Anal. Appl. Pyrolysis 2009, 86, 293–303. [Google Scholar] [CrossRef]
- Al-Salem, S.M.; Antelava, A.; Constantinou, A.; Manos, G.; Dutta, A. A review on thermal and catalytic pyrolysis of plastic solid waste (PSW). J. Environ. Manag. 2017, 197, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Sharuddin, S.D.A.; Abnisa, F.; Daud, W.; Aroua, M.K. A review on pyrolysis of plastic wastes. Energy Convers. Manag. 2016, 115, 308–326. [Google Scholar] [CrossRef]
- Zhang, F.; Zeng, M.; Yappert, R.D.; Sun, J.; Lee, Y.-H.; LaPointe, A.M.; Peters, B.; Abu-Omar, M.M.; Scott, S.L. Polyethylene upcycling to long-chain alkylaromatics by tandem hydrogenolysis/aromatization. Science 2020, 370, 437. [Google Scholar] [CrossRef]
- Lee, N.; Joo, J.; Lin, K.-Y.A.; Lee, J. Waste-to-Fuels: Pyrolysis of Low-Density Polyethylene Waste in the Presence of H-ZSM-11. Polymers 2021, 13, 1198. [Google Scholar] [CrossRef]
- Miandad, R.; Barakat, M.A.; Rehan, M.; Aburiazaiza, A.S.; Ismail, I.M.I.; Nizami, A.S. Plastic waste to liquid oil through catalytic pyrolysis using natural and synthetic zeolite catalysts. Waste Manag. 2017, 69, 66–78. [Google Scholar] [CrossRef]
- Mullen, C.A.; Dorado, C.; Boateng, A.A. Catalytic co-pyrolysis of switchgrass and polyethylene over HZSM-5: Catalyst deactivation and coke formation. J. Anal. Appl. Pyrolysis 2018, 129, 195–203. [Google Scholar] [CrossRef]
- Jia, X.; Qin, C.; Friedberger, T.; Guan, Z.; Huang, Z. Efficient and selective degradation of polyethylenes into liquid fuels and waxes under mild conditions. Sci. Adv. 2016, 2, 1501591. [Google Scholar] [CrossRef] [Green Version]
- Tennakoon, A.; Wu, X.; Paterson, A.L.; Patnaik, S.; Pei, Y.; LaPointe, A.M.; Ammal, S.C.; Hackler, R.A.; Heyden, A.; Slowing, I.I.; et al. Catalytic upcycling of high-density polyethylene via a processive mechanism. Nat. Catal. 2020, 3, 893–901. [Google Scholar] [CrossRef]
- Celik, G.; Kennedy, R.M.; Hackler, R.A.; Ferrandon, M.; Tennakoon, A.; Patnaik, S.; LaPointe, A.M.; Ammal, S.C.; Heyden, A.; Perras, F.A.; et al. Upcycling Single-Use Polyethylene into High-Quality Liquid Products. ACS Cent. Sci. 2019, 5, 1795–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Gu, J.; Wang, Y.; Zhang, J.; Yuan, H.; Chen, Y. Polyethylene high-pressure pyrolysis: Better product distribution and process mechanism analysis. Chem. Eng. J. 2020, 385, 123866. [Google Scholar] [CrossRef]
- Polotsky, A.A.; Kazakov, A.D.; Birshtein, T.M. Linear minority chain in a star brush: The coil-to-flower transition. Polymer 2017, 130, 242–249. [Google Scholar] [CrossRef]
- Li, B.; Kawakita, Y.; Ohira-Kawamura, S.; Sugahara, T.; Wang, H.; Wang, J.; Chen, Y.; Kawaguchi, S.I.; Kawaguchi, S.; Ohara, K.; et al. Colossal barocaloric effects in plastic crystals. Nature 2019, 567, 506–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cui, X.; Ge, H.; Yang, Y.; Wang, Y.; Zhang, C.; Li, J.; Deng, T.; Qin, Z.; Hou, X. Chemical Recycling of Carbon Fiber Reinforced Epoxy Resin Composites via Selective Cleavage of the Carbon–Nitrogen Bond. ACS Sustain. Chem. Eng. 2015, 3, 3332–3337. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, X.; Yang, Q.; Deng, T.; Wang, Y.; Yang, Y.; Jia, S.; Qin, Z.; Hou, X. Chemical recycling of unsaturated polyester resin and its composites via selective cleavage of the ester bond. Green Chem. 2015, 17, 4527–4532. [Google Scholar] [CrossRef]
- Sadykov, B.R.; Starikov, V.P.; Sadykov, R.K.; Kalabin, G.A. Determination of the fractional composition of merchantable oil using quantitative 1 H NMR spectra. Pet. Chem. 2012, 52, 22–27. [Google Scholar] [CrossRef]
- Morgenstern, M.; Cline, J.; Meyer, S.; Cataldo, S. Determination of the Kinetics of Biodiesel Production Using Proton Nuclear Magnetic Resonance Spectroscopy (1H NMR). Energy Fuels 2006, 20, 1350–1353. [Google Scholar] [CrossRef]
- Hu, Q.; Tang, Z.; Yao, D.; Yang, H.; Shao, J.; Chen, H. Thermal behavior, kinetics and gas evolution characteristics for the co-pyrolysis of real-world plastic and tyre wastes. J. Clean. Prod. 2020, 260, 121102. [Google Scholar] [CrossRef]
- Mazloum, S.; Awad, S.; Allam, N.; Aboumsallem, Y.; Loubar, K.; Tazerout, M. Modelling plastic heating and melting in a semi-batch pyrolysis reactor. Appl. Energy 2021, 283, 116375. [Google Scholar] [CrossRef]
- Breed, L.; Murrill, E. Solid-solid phase transitions determined by differential scanning calorimetry: Part I. Tetrahedral substances. Thermochim. Acta 1970, 1, 239–246. [Google Scholar]
- Chandra, D.; Ding, W.; Lynch, R.A.; Tomilinson, J.J. Phase transitions in “plastic crystals”. J. Less Common Met. 1991, 168, 159–167. [Google Scholar] [CrossRef]
- Zhang, Y.; Duan, D.; Lei, H.; Villota, E.; Ruan, R. Jet fuel production from waste plastics via catalytic pyrolysis with activated carbons. Appl. Energy 2019, 251, 113337. [Google Scholar] [CrossRef]
- Zhao, D.; Wang, X.; Miller, J.B.; Huber, G.W. The Chemistry and Kinetics of Polyethylene Pyrolysis: A Process to Produce Fuels and Chemicals. ChemSusChem 2020, 13, 1764–1774. [Google Scholar] [CrossRef]
- Kumari, A.; Kumar, S. Pyrolytic degradation of polyethylene in autoclave under high pressure to obtain fuel. J. Anal. Appl. Pyrolysis 2017, 124, 298–302. [Google Scholar] [CrossRef]
- Ding, K.; Liu, S.; Huang, Y.; Liu, S.; Zhou, N.; Peng, P.; Wang, Y.; Chen, P.; Ruan, R. Catalytic microwave-assisted pyrolysis of plastic waste over NiO and HY for gasoline-range hydrocarbons production. Energy Convers. Manag. 2019, 196, 1316–1325. [Google Scholar] [CrossRef]
- Sirota, E.B.; Singer, D.M.; King, H.E. Structural effects of high pressure gas on the rotator phases of normal alkanes. J. Chem. Phys. 1994, 100, 1542–1551. [Google Scholar] [CrossRef]
- Mohanraj, C.; Senthilkumar, T.; Chandrasekar, M. A review on conversion techniques of liquid fuel from waste plastic materials. Int. J. Energy Res. 2017, 41, 1534–1552. [Google Scholar]
- Jia, C.; Xie, S.; Zhang, W.; Intan, N.N.; Sampath, J.; Pfaendtner, J.; Lin, H. Deconstruction of high-density polyethylene into liquid hydrocarbon fuels and lubricants by hydrogenolysis over Ru catalyst. Chem Catal. 2021, 1, 437–455. [Google Scholar] [CrossRef]
- Shi, S. Advances in modeling hydrocarbon cracking kinetic predictions by quantum chemical theory: A review. Int. J. Energy Res. 2018, 42, 3164–3181. [Google Scholar] [CrossRef]
- Jehanno, C.; Alty, J.W.; Roosen, M.; De Meester, S.; Dove, A.P.; Chen, E.Y.; Leibfarth, F.A.; Sardon, H. Critical advances and future opportunities in upcycling commodity polymers. Nature 2022, 603, 803–814. [Google Scholar] [CrossRef]
- Chen, H.; Wan, K.; Zhang, Y.; Wang, Y. Waste to Wealth: Chemical Recycling and Chemical Upcycling of Waste Plastics for a Great Future. ChemSusChem 2021, 14, 4123–4136. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Reactants | Reactant Mass (g) | Set Temperature (°C) | Initial Pressure (Bar) | Atmosphere |
|---|---|---|---|---|---|
| Blank | - | - | 380 | 21 | Nitrogen |
| P1 | LDPE | 11 | 380 | 1 atm | Nitrogen |
| P2 | LDPE | 11 | 380 | 2 | Nitrogen |
| P3 | LDPE | 11 | 380 | 3 | Nitrogen |
| P4 | LDPE | 11 | 380 | 4 | Nitrogen |
| P5 | LDPE | 11 | 380 | 5 | Nitrogen |
| P6 | LDPE | 11 | 380 | 6 | Nitrogen |
| P11 | LDPE | 11 | 380 | 11 | Nitrogen |
| P16 | LDPE | 11 | 380 | 16 | Nitrogen |
| P21 | LDPE | 11 | 380 | 21 | Nitrogen |
| A1 | LDPE | 11 | 380 | 21 | Helium |
| A2 | LDPE | 11 | 380 | 21 | Argon |
| L1 | LDPE + 1-hexene | 8 + 8 | 380 | 1 atm | Nitrogen |
| L2 | LDPE + 1-octene | 8 + 8 | 380 | 1 atm | Nitrogen |
| L3 | LDPE + 1-decene | 8 + 8 | 380 | 1 atm | Nitrogen |
| L4 | LDPE + 1-hexene | 8 + 3 | 380 | 21 | Nitrogen |
| L5 | LDPE + 1-octene | 8 + 3 | 380 | 21 | Nitrogen |
| L6 | LDPE + 1-decene | 8 + 3 | 380 | 21 | Nitrogen |
| L7 | LDPE + octane | 8 + 8 | 380 | 1 atm | Nitrogen |
| L8 | LDPE + xylene | 8 + 8 | 380 | 1 atm | Nitrogen |
| L9 | LDPE + octane | 8 + 3 | 380 | 21 | Nitrogen |
| L10 | LDPE + xylene | 8 + 3 | 380 | 21 | Nitrogen |
| L11 | LDPE + 1-hexene | 8 + 8 | 340 | 1 atm | Nitrogen |
| Name | Gaseous Components (mg/g LDPE) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P5 | P6 | P11 | P16 | P21 | |
| Hydrogen | 0.00 | 0.00 | 0.11 | 0.10 | 0.15 | 0.19 | 0.23 | 0.25 | 0.27 |
| Methane | 0.21 | 0.07 | 2.77 | 2.89 | 3.72 | 4.34 | 5.55 | 6.34 | 6.99 |
| Ethane | 0.24 | 0.28 | 4.06 | 4.39 | 5.15 | 5.98 | 8.79 | 10.34 | 11.65 |
| Ethylene | 0.09 | 0.09 | 1.54 | 1.48 | 1.70 | 1.90 | 2.13 | 2.03 | 1.91 |
| Propane | 0.30 | 0.42 | 4.37 | 4.87 | 4.62 | 5.19 | 8.68 | 10.66 | 11.55 |
| Propene | 0.17 | 0.33 | 3.83 | 4.08 | 3.93 | 4.35 | 6.34 | 6.87 | 6.68 |
| C4 alkanes | 0.15 | 0.22 | 1.89 | 2.27 | 1.85 | 2.12 | 3.57 | 4.90 | 4.82 |
| C4 olefins | 0.14 | 0.17 | 0.19 | 0.24 | 1.35 | 1.47 | 2.14 | 2.54 | 2.24 |
| C5 | 0.07 | 0.17 | 1.05 | 1.42 | 1.25 | 1.67 | 2.68 | 3.23 | 3.63 |
| C6 | 0.35 | 0.37 | 0.77 | 0.82 | 1.00 | 0.47 | 0.89 | 1.01 | 0.94 |
| Sum | 1.73 | 2.12 | 20.58 | 22.57 | 24.72 | 27.68 | 41.00 | 48.18 | 50.67 |
| Components | Peak Area % | |||||||
|---|---|---|---|---|---|---|---|---|
| P2 | P3 | P4 | P5 | P6 | P11 | P16 | P21 | |
| n-paraffins | 68.46 | 67.37 | 67.48 | 63.88 | 62.76 | 60.83 | 59.17 | 59.56 |
| C4 | 1.91 | 1.45 | 1.34 | 1.19 | 1.59 | 1.08 | 2.25 | 0.77 |
| C5 | 3.86 | 3.25 | 3.21 | 3.14 | 3.89 | 3.14 | 4.81 | 3.12 |
| C6 | 3.68 | 3.28 | 3.41 | 3.23 | 3.64 | 3.59 | 4.58 | 5.98 |
| C7 | 3.16 | 3.07 | 3.29 | 4.03 | 4.24 | 4.52 | 4.43 | 4.87 |
| C8 | 3.05 | 3.10 | 3.31 | 3.83 | 4.09 | 4.34 | 4.44 | 4.68 |
| C9 | 2.71 | 2.85 | 2.99 | 3.34 | 3.61 | 3.85 | 3.79 | 4.08 |
| C10 | 2.71 | 2.93 | 2.94 | 3.20 | 3.49 | 3.66 | 3.61 | 3.89 |
| C11 | 2.70 | 3.06 | 2.95 | 3.17 | 3.43 | 3.66 | 3.48 | 3.75 |
| C12 | 2.56 | 2.93 | 2.79 | 3.09 | 3.30 | 3.38 | 3.32 | 3.61 |
| C13 | 2.39 | 2.67 | 2.57 | 2.85 | 2.82 | 3.02 | 2.75 | 3.00 |
| C14 | 2.24 | 2.45 | 2.39 | 2.53 | 2.61 | 2.63 | 2.50 | 2.62 |
| C15 | 2.62 | 2.86 | 2.77 | 2.75 | 2.79 | 2.83 | 2.65 | 2.74 |
| C15+ | 34.85 | 33.47 | 33.53 | 27.52 | 23.26 | 21.14 | 16.57 | 16.46 |
| Isoparaffins | 1.37 | 2.13 | 1.69 | 1.96 | 1.73 | 1.79 | 2.02 | 2.38 |
| Cycloparaffins | 4.05 | 4.05 | 4.48 | 5.48 | 5.90 | 7.26 | 7.89 | 8.38 |
| Cyclopentane, methyl- | 0.44 | 0.41 | 0.44 | 0.58 | 0.65 | 0.72 | 0.89 | 0.84 |
| Cyclohexane, methyl- | 0.75 | 0.74 | 0.78 | 0.95 | 1.03 | 1.12 | 1.22 | 1.26 |
| Cyclopentane, ethyl- | 0.24 | 0.25 | 0.26 | 0.33 | 0.37 | 0.41 | 0.46 | 0.48 |
| Cyclohexane, ethyl- | 0.36 | 0.36 | 0.37 | 0.45 | 0.50 | 0.56 | 0.59 | 0.63 |
| Others | 2.26 | 2.29 | 2.62 | 3.16 | 3.37 | 4.44 | 4.73 | 5.17 |
| α-Olefins | 14.99 | 14.85 | 14.62 | 15.41 | 15.68 | 15.78 | 15.50 | 13.67 |
| C6 | 2.68 | 2.41 | 2.29 | 2.35 | 2.56 | 2.15 | 2.42 | 2.06 |
| C7 | 1.87 | 1.74 | 1.80 | 2.18 | 2.25 | 2.19 | 2.20 | 2.28 |
| C8 | 2.08 | 2.05 | 2.11 | 2.35 | 2.40 | 2.38 | 2.27 | 1.60 |
| C9 | 1.27 | 1.22 | 1.32 | 1.42 | 1.52 | 1.60 | 1.50 | 1.29 |
| C10 | 1.47 | 1.58 | 1.50 | 1.52 | 1.59 | 1.34 | 1.23 | 1.24 |
| C10+ | 5.63 | 5.84 | 5.60 | 5.59 | 5.36 | 6.12 | 5.88 | 5.20 |
| Cycloalkenes | 1.60 | 1.64 | 1.65 | 2.15 | 2.49 | 2.60 | 2.70 | 2.77 |
| Other olefins | 5.27 | 5.34 | 5.53 | 6.90 | 4.33 | 6.45 | 6.56 | 6.32 |
| Aromatics | - | - | - | 0.31 | 0.34 | 1.19 | 1.29 | 1.53 |
| Benzene | - | - | - | - | - | - | - | - |
| Toluene | - | - | - | 0.31 | 0.34 | 0.53 | 0.61 | 0.75 |
| Xylene | - | - | - | - | - | - | - | - |
| Other MAHs | - | - | - | - | - | 0.65 | 0.68 | 0.78 |
| Others | 4.26 | 4.63 | 4.54 | 3.90 | 4.33 | 4.10 | 4.87 | 5.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Chen, X.; Cheng, L.; Gu, J.; Xu, Y. Conversion of Polyethylene to High-Yield Fuel Oil at Low Temperatures and Atmospheric Initial Pressure. Int. J. Environ. Res. Public Health 2023, 20, 4048. https://doi.org/10.3390/ijerph20054048
Zhang Y, Chen X, Cheng L, Gu J, Xu Y. Conversion of Polyethylene to High-Yield Fuel Oil at Low Temperatures and Atmospheric Initial Pressure. International Journal of Environmental Research and Public Health. 2023; 20(5):4048. https://doi.org/10.3390/ijerph20054048
Chicago/Turabian StyleZhang, Yuanjia, Xueru Chen, Leilei Cheng, Jing Gu, and Yulin Xu. 2023. "Conversion of Polyethylene to High-Yield Fuel Oil at Low Temperatures and Atmospheric Initial Pressure" International Journal of Environmental Research and Public Health 20, no. 5: 4048. https://doi.org/10.3390/ijerph20054048