
Epithelial Ablation of Miro1/Rhot1 GTPase Augments Lung Inflammation by Cigarette Smoke



Abstract
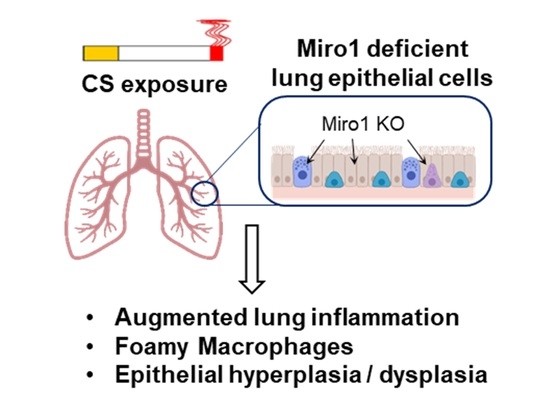
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Model and Exposure
2.2. Cigarette Smoke Exposure
2.3. Collection of Bronchoalveolar Lavage (BAL)
2.4. Total Cell Count in BAL Fluid
2.5. Differential Cell Count in BAL Fluid
2.6. Inflammatory Mediators
2.7. Measurement of Lung Mechanics
2.8. Lung Morphometry and Histopathology
2.9. Statistical Analysis
3. Results
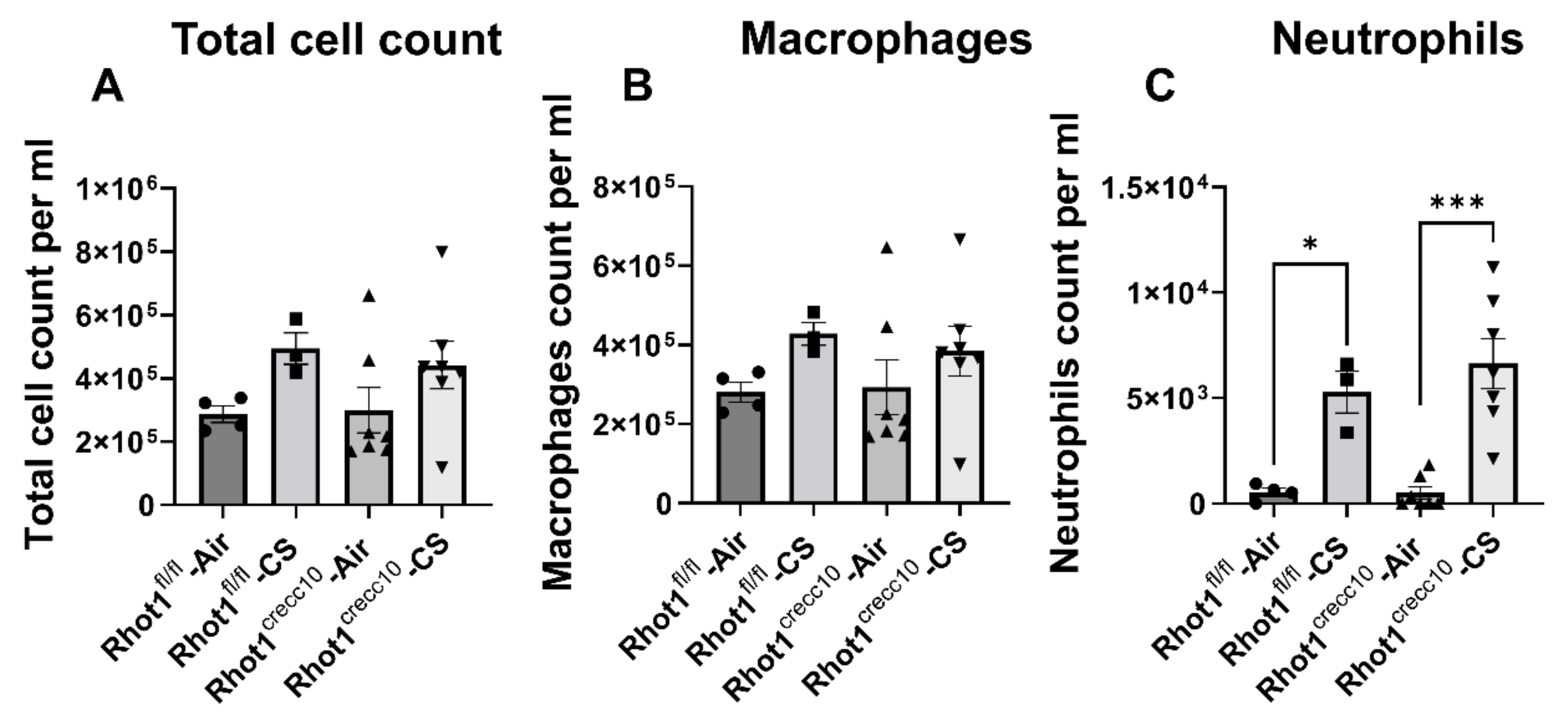
3.1. Three Days (Acute) Exposure
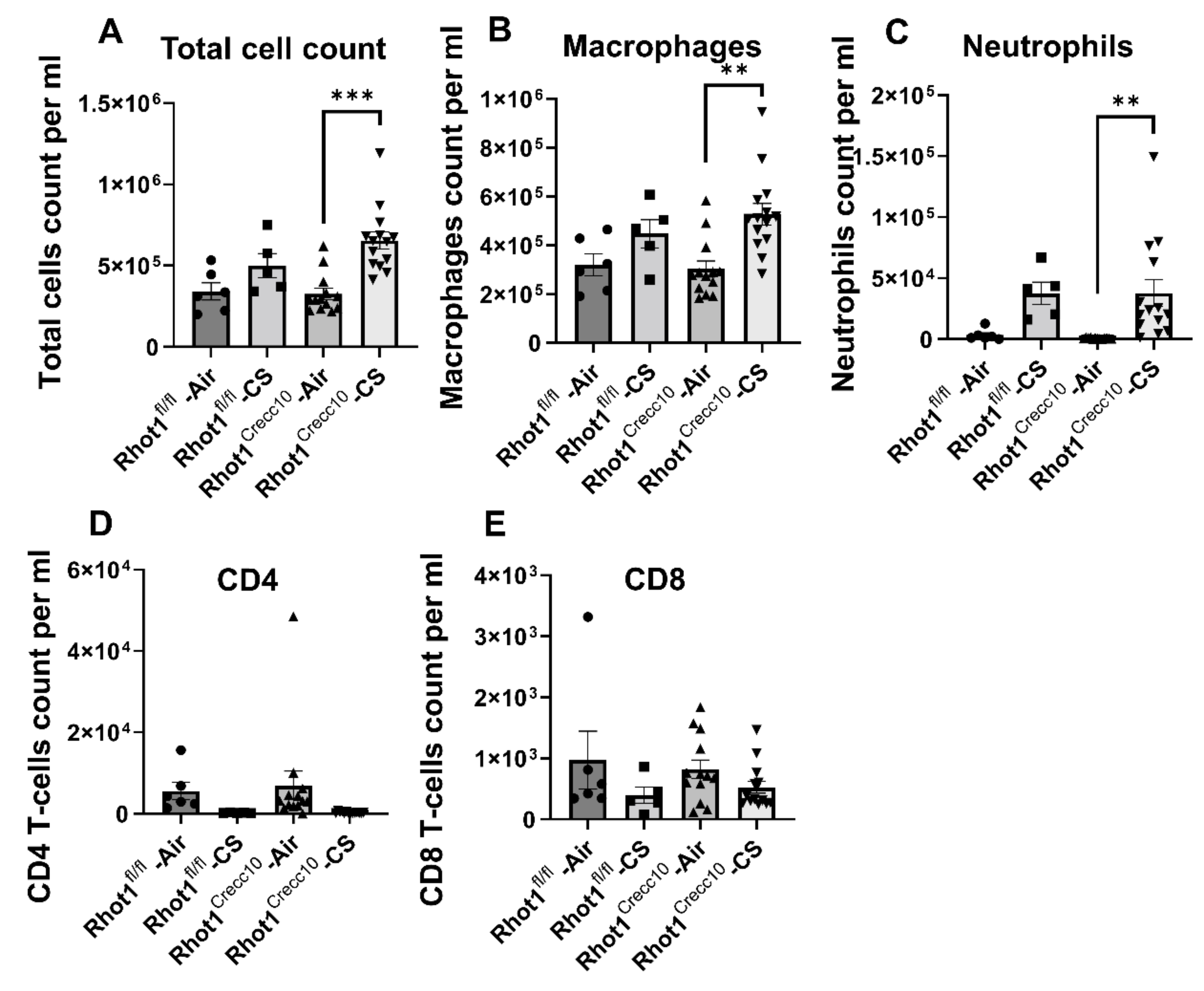
3.1.1. Inflammatory Cellular Influx in Rhot1 Epithelial Cell Specific KO (Rhot1CreCC10) and WT (Rhot1fl/fl) Mice
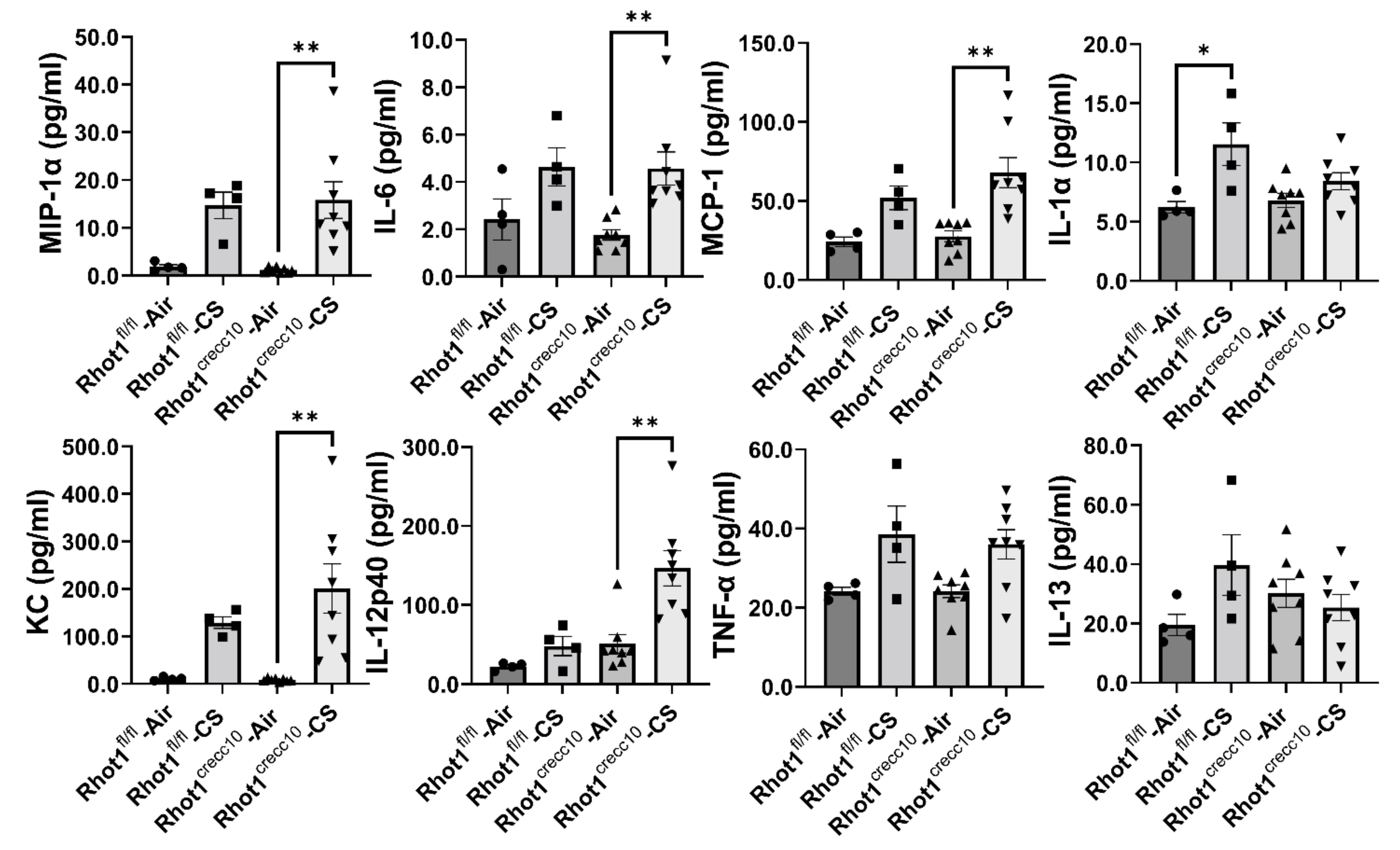
3.1.2. Effect of Acute, CS Exposure on Levels of Pro-Inflammatory Mediators in Rhot1 Epithelial Cell Specific KO (Rhot1CreCC10) and WT (Rhot1fl/fl) Mice
3.2. Four Months (Chronic) Exposure
3.2.1. Inflammatory Cellular Influx in Rhot1 Epithelial Cell Specific KO (Rhot1CreCC10) and WT (Rhot1fl/fl) Mice by Chronic CS Exposures
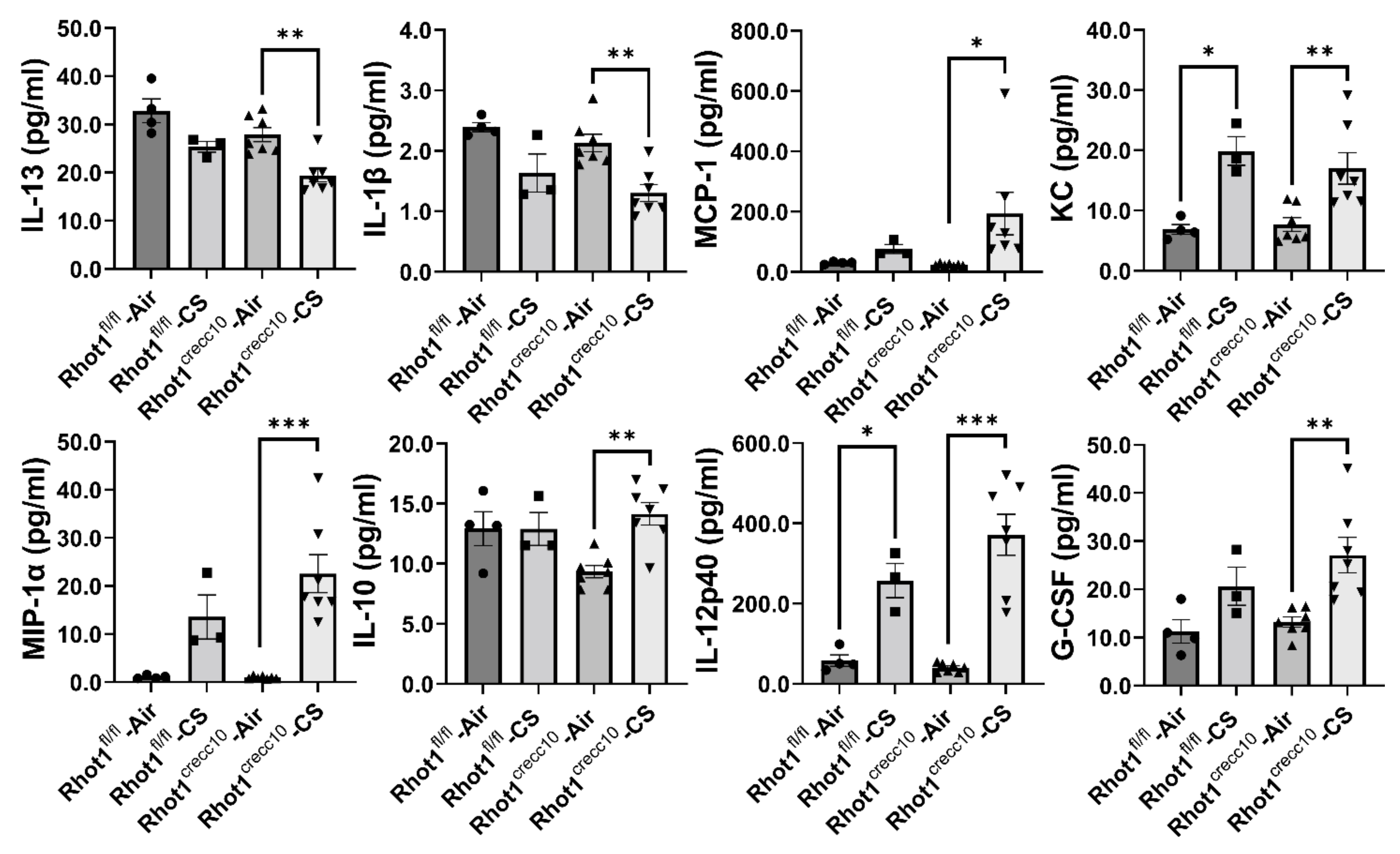
3.2.2. Effect of Chronic, CS Exposure on Levels of Pro-Inflammatory Mediators in Rhot1 Epithelial Cell Specific KO (Rhot1CreCC10) and WT (Rhot1fl/fl) Mice
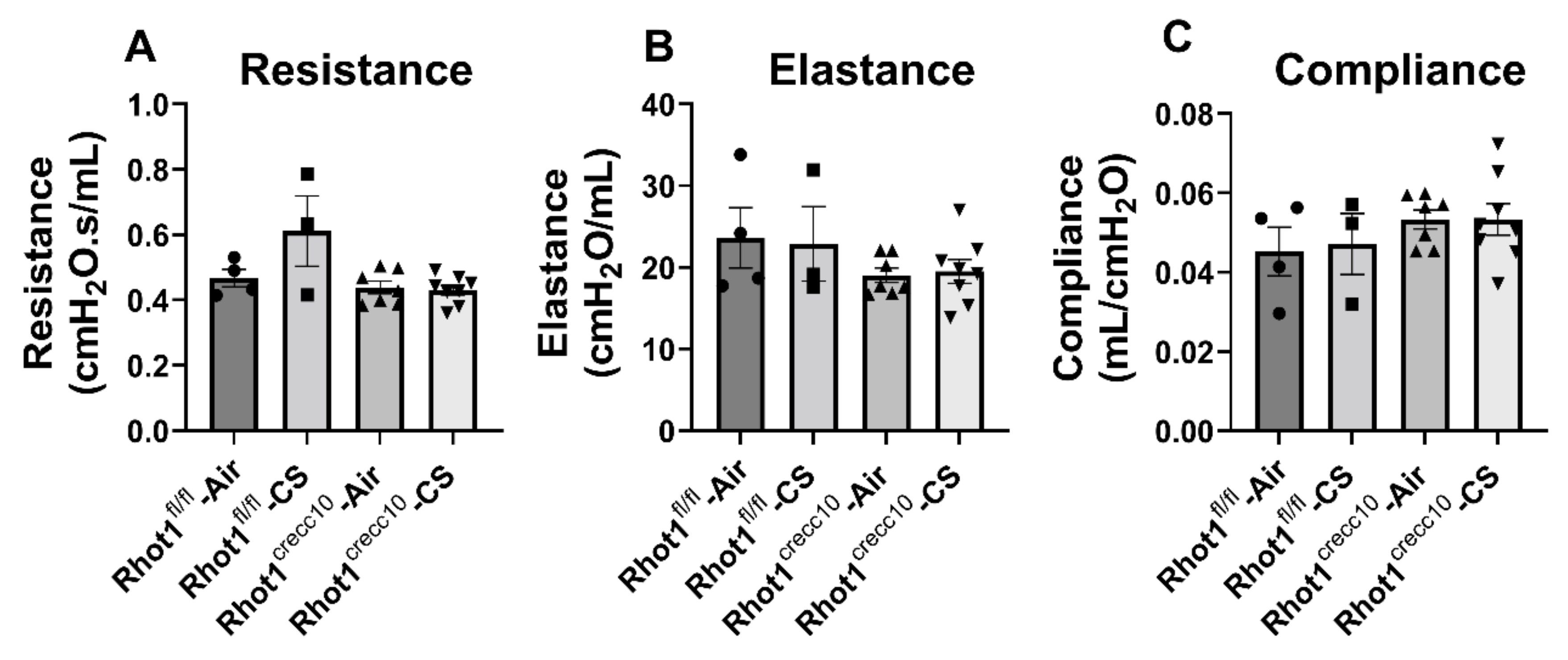
3.2.3. Effect of Chronic CS on Lung Mechanical Properties in Rhot1 Epithelial Cell Specific KO (Rhot1CreCC10) WT (Rhot1fl/fl) Mice
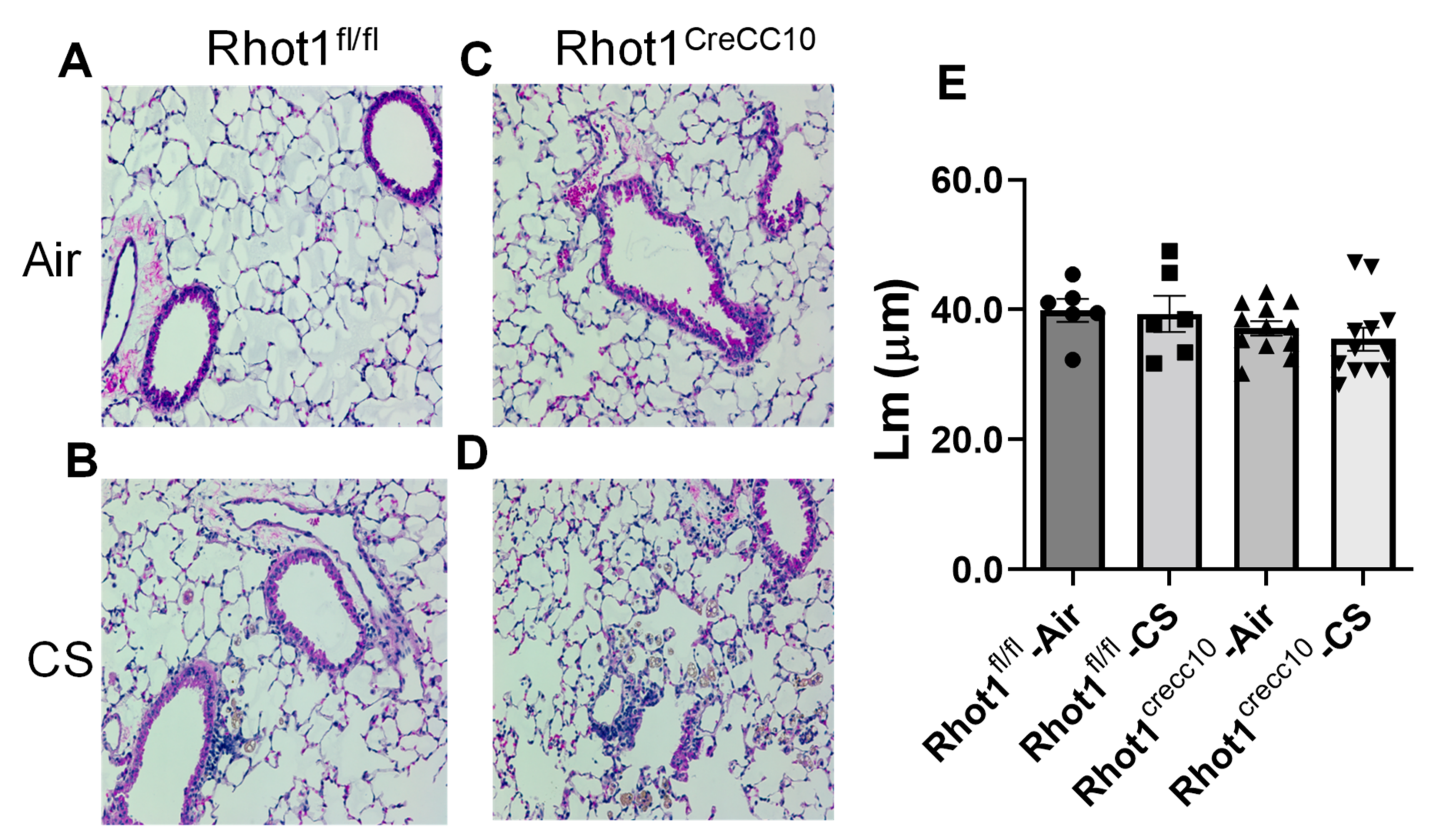
3.2.4. Effect on Airspace Enlargement and Histopathology of Lung Tissues in Rhot1 Epithelial Cell Specific KO (Rhot1CreCC10) and WT (Rhot1fl/fl) Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sundar, I.K.; Maremanda, K.; Rahman, I. Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke. Toxicol. Lett. 2019, 317, 92–101. [Google Scholar] [CrossRef]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshaabi, H.; Shannon, N.; Gravelle, R.; Milczarek, S.; Messier, T.; Cunniff, B. Miro1-mediated mitochondrial positioning supports subcellular redox status. Redox Biol. 2021, 38, 101818. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, C.A.; Sundar, I.K.; Rahman, I. Mitochondrial redox system, dynamics, and dysfunction in lung inflammaging and COPD. Int. J. Biochem. Cell Biol. 2016, 81, 294–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix CM, S.; Kamga, C.; Lee, J.S. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [Green Version]
- Safiulina, D.; Kuum, M.; Choubey, V.; Gogichaishvili, N.; Liiv, J.; Hickey, M.A.; Liiv, M. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 2019, 38, e99384. [Google Scholar] [CrossRef] [PubMed]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.H.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemani, N.; Carvalho, E.; Tomar, D.; Dong, Z.; Ketschek, A.; Breves, S.L.; Palaniappan, P. MIRO-1 determines mitochondrial shape transition upon GPCR activation and Ca2+ stress. Cell Rep. 2018, 23, 1005–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Dai, L.; Wang, T.; He, J.; Wang, Y.; Wen, F. The elevated CXCL5 levels in circulation are associated with lung function decline in COPD patients and cigarette smoking-induced mouse model of COPD. Ann. Med. 2019, 51, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Chung, S.; Hwang, J.-W.; Arunachalam, G.; Cook, S.; Yao, H.; Rahman, I. Peroxiredoxin 6 differentially regulates acute and chronic cigarette smoke mediated lung inflammatory response and injury. Exp. Lung Res. 2010, 36, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Xu, D.; Wang, T.; Yuan, Z.; Liu, L.; Shen, Y.; Wen, F. Mitoquinone ameliorates cigarette smoke-induced airway inflammation and mucus hypersecretion in mice. Int. Immunopharmacol. 2021, 90, 107149. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, G.; Sundar, I.K.; Hwang, J.-W.; Yao, H.; Rahman, I. Emphysema is associated with increased inflammation in lungs of atherosclerosis-prone mice by cigarette smoke: Implications in comorbidities of COPD. J. Inflamm. 2010, 7, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Rajendrasozhan, S.; Yao, H.; Chung, S.; Sundar, I.K.; Huyck, H.L.; Rahman, I. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J. Immunol. 2011, 187, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Lerner, C.A.; Lei, W.; Sundar, I.K.; Rahman, I. Genetic Ablation of CXCR2 Protects against Cigarette Smoke-Induced Lung Inflammation and Injury. Front. Pharmacol. 2016, 7, 391. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Edirisinghe, I.; Rajendrasozhan, S.; Yang, S.-R.; Caito, S.; Adenuga, D.; Rahman, I. Cigarette smoke-mediated inflammatory and oxidative responses are strain-dependent in mice. Am. J. Physiol. Cell. Mol. Physiol. 2008, 294, L1174–L1186. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, T.; Shen, Y.; Xu, D.; Li, X.; An, J.; Chen, L. Knockout of RAGE ameliorates mainstream cigarette smoke-induced airway inflammation in mice. Int. Immunopharmacol. 2017, 50, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Rajendrasozhan, S.; Chung, S.; Sundar, I.K.; Yao, H.; Rahman, I. Targeted disruption of NF-κB1 (p50) augments cigarette smoke-induced lung inflammation and emphysema in mice: A critical role of p50 in chromatin remodeling. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L197–L209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, T.T.; Winden, D.R.; Marlor, D.R.; Wright, A.J.; Jones, C.M.; Chavarria, M.; Reynolds, P.R. Acute secondhand smoke-induced pulmonary inflammation is diminished in RAGE knockout mice. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2014, 307, L758–L764. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Liu, K.W.K.; Ip, M.S.M.; Liang, Y.; Mak, J.C.W. Protective effect of selegiline on cigarette smoke-induced oxidative stress and inflammation in rat lungs in vivo. Ann. Transl. Med. 2020, 8, 1418. [Google Scholar] [CrossRef]
- Lim, D.; Kim, W.; Lee, C.; Bae, H.; Kim, J. Macrophage Depletion Protects against Cigarette Smoke-Induced Inflammatory Response in the Mouse Colon and Lung. Front. Physiol. 2018, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Phillips, B.; Veljkovic, E.; Peck, M.J.; Buettner, A.; Elamin, A.; Guedj, E.; Bou, S. A 7-month cigarette smoke inhalation study in C57BL/6 mice demonstrates reduced lung inflammation and emphysema following smoking cessation or aerosol exposure from a prototypic modified risk tobacco product. Food Chem. Toxicol. 2015, 80, 328–345. [Google Scholar] [CrossRef]
- Yao, H.; Chung, S.; Hwang, J.; Rajendrasozhan, S.; Sundar, I.K.; Dean, D.A.; Rnty, M. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Investig. 2012, 122, 2032–2045. [Google Scholar] [CrossRef]
- Nyunoya, T.; Mebratu, Y.; Contreras, A.; Delgado, M.; Chand, H.S.; Tesfaigzi, Y. Molecular Processes that Drive Cigarette Smoke–Induced Epithelial Cell Fate of the Lung. Am. J. Respir. Cell Mol. Biol. 2014, 50, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Fujii, S. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef]
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, L840–L854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballweg, K.; Mutze, K.; Königshoff, M.; Eickelberg, O.; Meiners, S. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L895–L907. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Numata, T. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef] [Green Version]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Washko, G.R. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Kanerva, J. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dorn, G.W., II. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Fandos, D.; Peinado, V.I.; Puig-Pey, R.; Ferrer, E.; Musri, M.M.; Ramírez, J.; Barberà, J.A. Pulmonary Inflammatory Reaction and Structural Changes Induced by Cigarette Smoke Exposure in the Guinea Pig. J. Chronic Obstr. Pulm. Dis. 2012, 9, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Gorska, K.; Krenke, R.; Korczynski, P.; Kosciuch, J.; Domagala-Kulawik, J.; Chazan, R. Eosinophilic airway inflammation in chronic obstructive pulmonary disease and asthma. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2008, 59, 261–270. [Google Scholar]
- Klosowiak, J.L.; Park, S.; Smith, K.P.; French, M.E.; Focia, P.J.; Freymann, D.M.; Rice, S.E. Structural insights into Parkin substrate lysine targeting from minimal Miro substrates. Sci. Rep. 2016, 6, 33019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Deak, M. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Domenech, G.; Covill-Cooke, C.; Howden, J.H.; Birsa, N.; Morfill, C.; Brandon, N.J.; Kittler, J.T. Miro ubiquitination is critical for efficient damage-induced PINK1/Parkin-mediated mitophagy. bioRxiv 2018. [Google Scholar] [CrossRef]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T.; Kurita, Y. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Wang, Q.; Muthumalage, T.; Rahman, I. Epithelial Ablation of Miro1/Rhot1 GTPase Augments Lung Inflammation by Cigarette Smoke. Pathophysiology 2021, 28, 501-512. https://doi.org/10.3390/pathophysiology28040033
Sharma S, Wang Q, Muthumalage T, Rahman I. Epithelial Ablation of Miro1/Rhot1 GTPase Augments Lung Inflammation by Cigarette Smoke. Pathophysiology. 2021; 28(4):501-512. https://doi.org/10.3390/pathophysiology28040033
Chicago/Turabian StyleSharma, Shikha, Qixin Wang, Thivanka Muthumalage, and Irfan Rahman. 2021. "Epithelial Ablation of Miro1/Rhot1 GTPase Augments Lung Inflammation by Cigarette Smoke" Pathophysiology 28, no. 4: 501-512. https://doi.org/10.3390/pathophysiology28040033
APA StyleSharma, S., Wang, Q., Muthumalage, T., & Rahman, I. (2021). Epithelial Ablation of Miro1/Rhot1 GTPase Augments Lung Inflammation by Cigarette Smoke. Pathophysiology, 28(4), 501-512. https://doi.org/10.3390/pathophysiology28040033