
The Role of Exposomes in the Pathophysiology of Autoimmune Diseases I: Toxic Chemicals and Food



Abstract
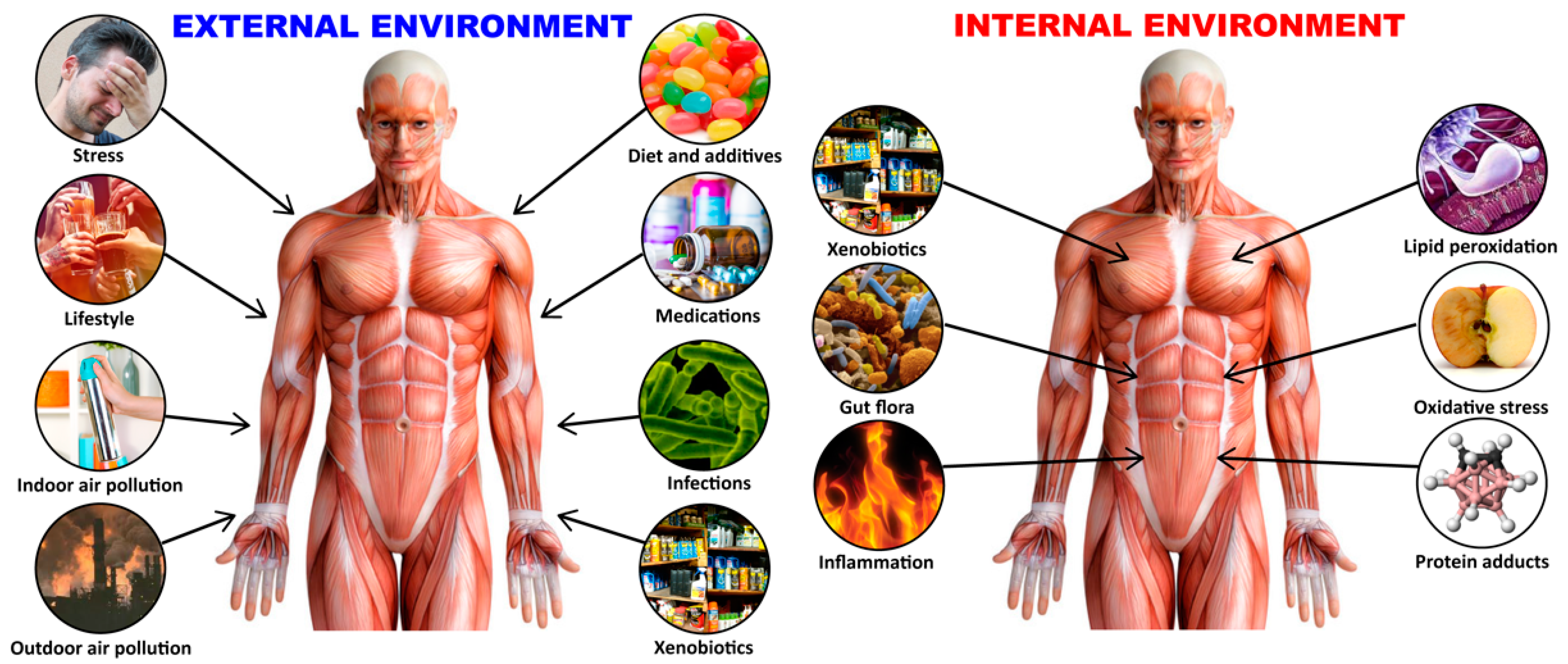
1. Introduction
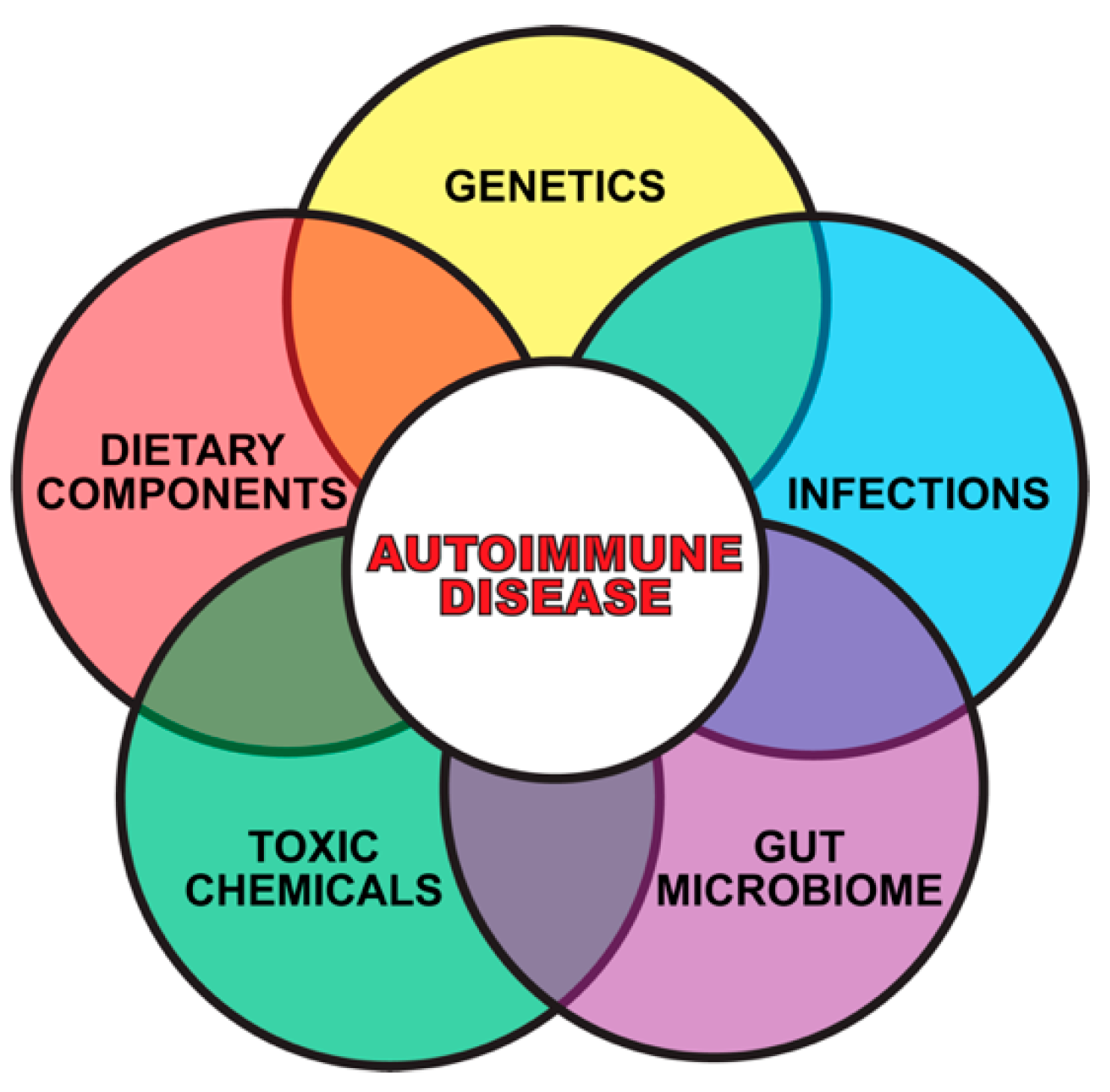
2. Gene–Environment Interaction in Autoimmune Diseases
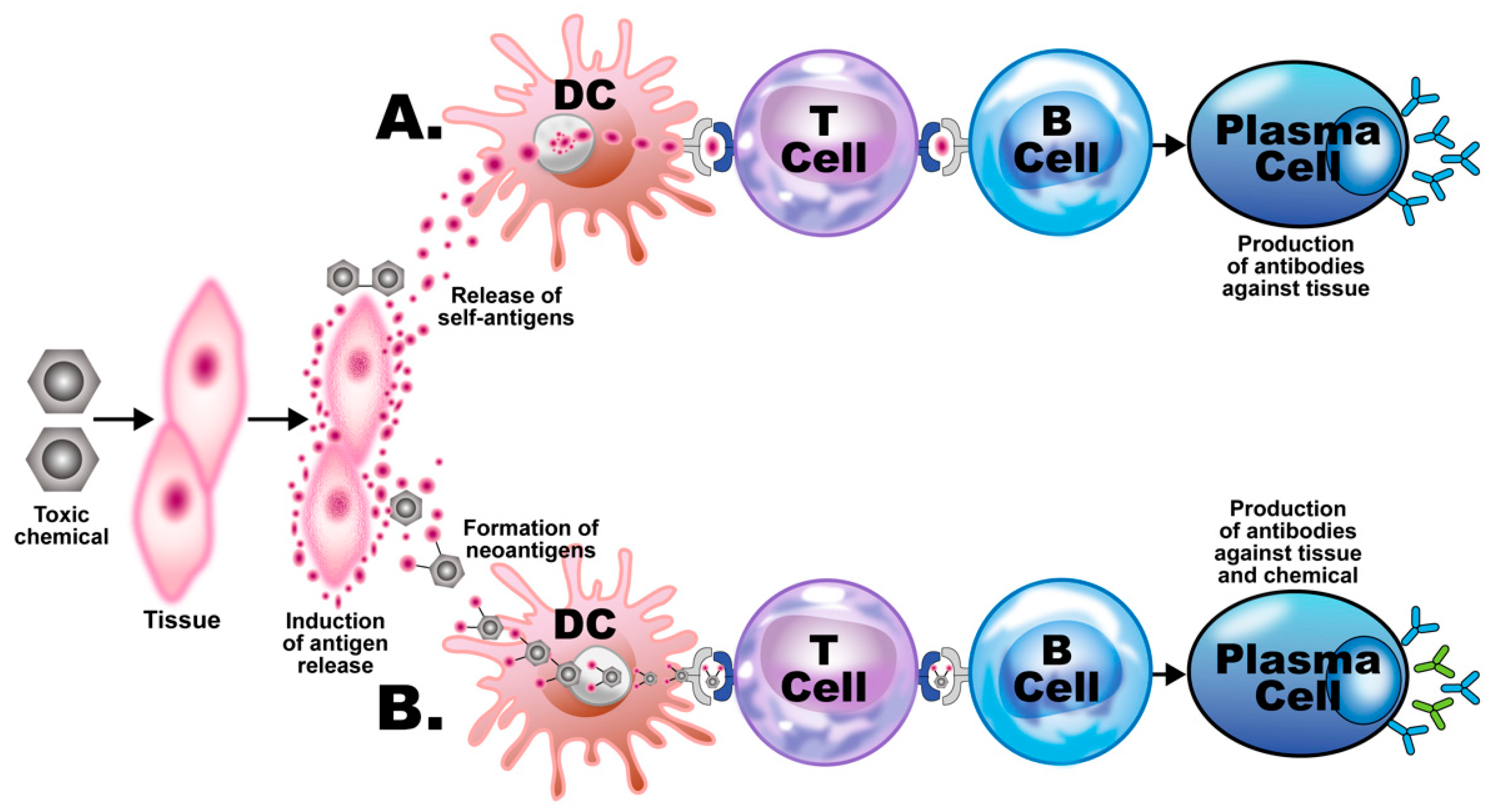
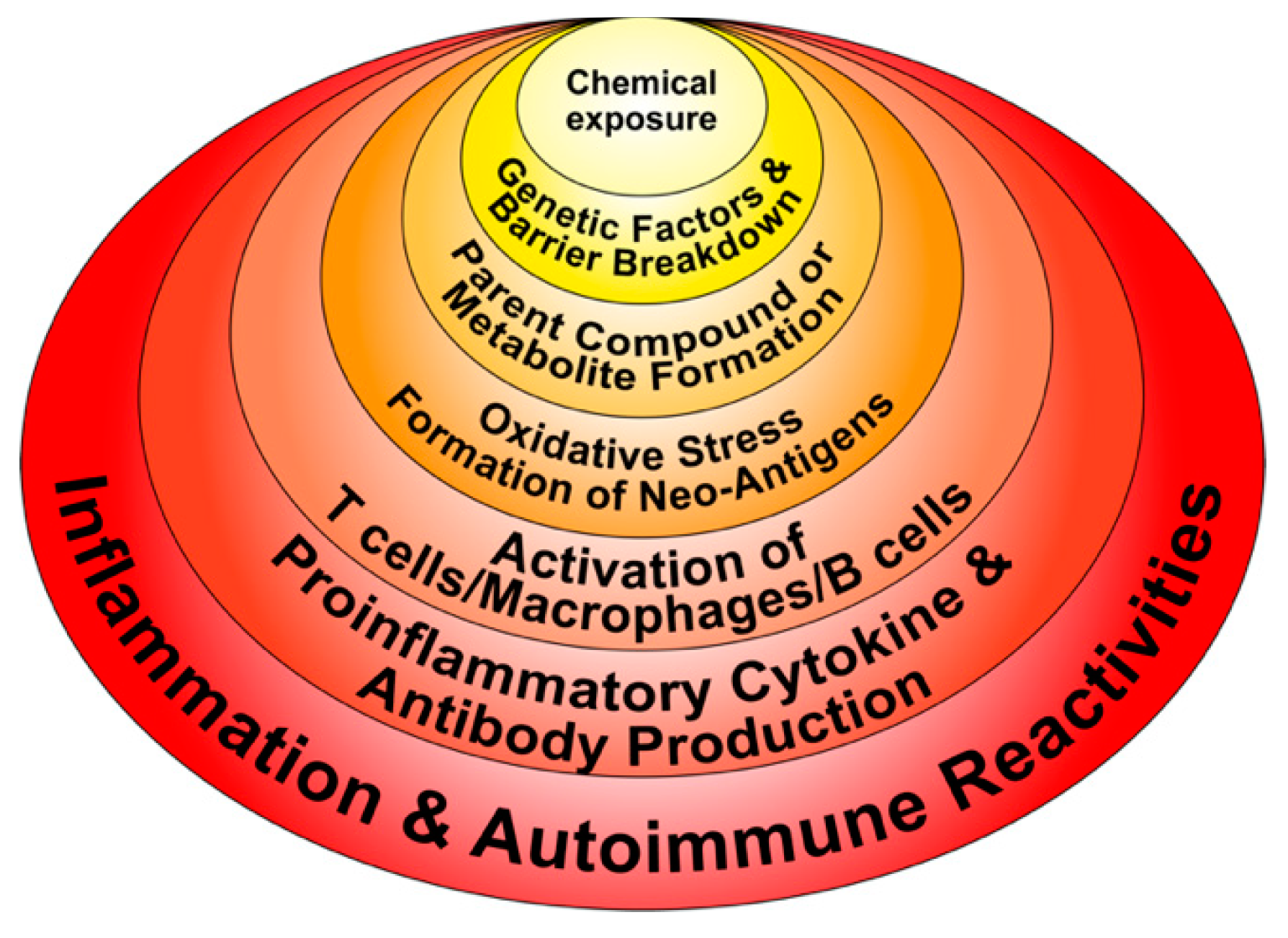
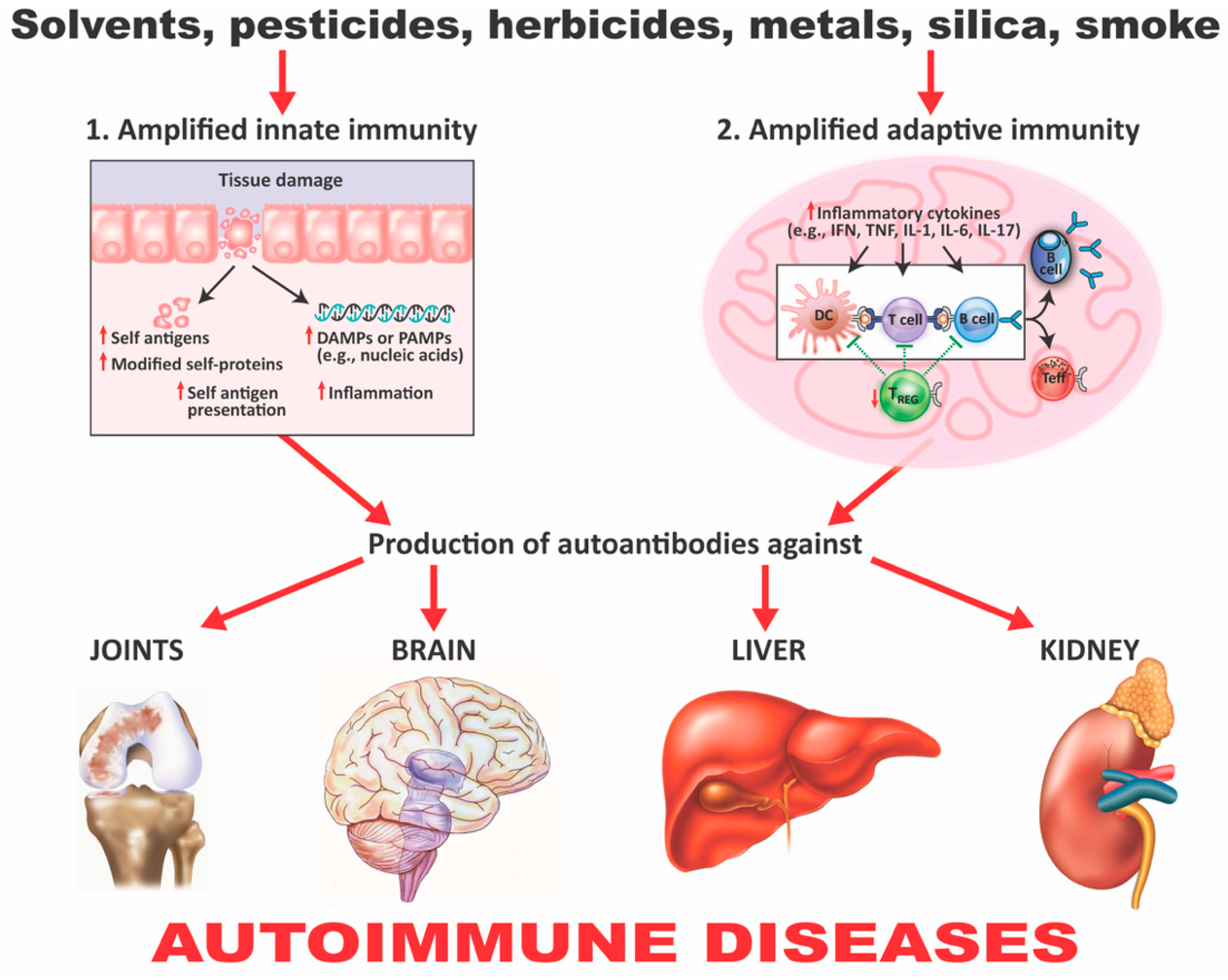
3. The Role of Toxic Chemicals in the Pathophysiology of Autoimmune Diseases
- Chemicals can alter cellular proliferation, Th1, Th2, Th3, Th17, apoptosis, and tissue-specific function;
- Chemicals can induce protein or lipid adducts, which activate Th17 cells and induce the production of IL-17 and IL-21;
- Chemicals can activate HSP90, inducing production of anti-HSP90 autoantibodies;
- Chemicals can increase reactive oxygen species (ROS) production and the induction of DNA fragmentation;
- Chemicals could interfere with iodine transportation or compete with thyroid hormones, inducing oxidative stress that leads to an inflammatory response by the thyroid gland;
- Chemicals not only stimulate the release of ROS, but also stimulate the synthesis of nitric oxide by nitric oxide synthase;
- Chemicals and environmental triggers in general can modify DNA methylation, inducing changes in gene expression. For example, alcohol consumption, smoking cigarettes, and exposure to environmental pollutants have been associated with autoimmunity induction through the induction of DNA methylation [51,52,53,54].
Mercury-Induced Autoimmunity
- Mercury-induced proliferation of human lymphocytes has been shown to occur 6 days postexposure with increased expression of several cytokines, including TNF-α, IL-1β, IL-6, and IL-8 in peripheral blood mononuclear cells (PBMCs). This lymphoproliferative response drives Th2 cell response [65];
- Very low (micromolar) concentration of mercuric chloride (HgCl2) can negatively affect the function of neutrophils; this is demonstrated by the enhanced production of hydrogen peroxide (H2O2), increased lysosomal enzymes, and the formation of neutrophil extracellular traps. These findings indicate the involvement of these cells in local tissue injury induced by mercury [66];
- In epidemiological studies, elevated levels of the inflammatory markers IFN-γ, TNF-α, and IL-1β were found in the sera of Amazonian gold miners in Brazil. Mercury was used to recover minute pieces of gold. Fish consumers from the same place who were exposed to mercury also showed increased levels of IFN-γ, IL-4, IL-6, and IL-17 cytokines [67];
- Mercury exposure is associated with the production of autoantibodies. In the same epidemiological study described above [67], autoantibodies were detected in the artisanal Amazonian gold miners. A positive correlation also was shown between the consumption of fish by the Amazonians and the presence of antinuclear antibodies (ANA) [67,68]. This same positive correlation was shown between fish consumption by members of the Cheyenne River Sioux tribe of the female gender and the presence of ANA in their blood [69]. Mercury was also detected in the blood of Faroese children and associated with multiple neural and non-neural IgM antibodies.
- 5.
- Mercury-induced nephrotic syndrome is an established outcome of mercury exposure in humans [72] through such things as mercury-containing cosmetics, hair dyes, mercury-containing pills, and occupational contact [73,74,75]. A review of the literature found that out of 26 renal biopsy cases, 21 had glomerular diseases, with the major pathological observations being membranous glomerulonephritis (15 patients) and minimal change disease (4 patients). Immune complexes and autoantibodies have been found in some patients, but not in others [72,73,74,75,76]. The mechanisms that lead to mercury-induced glomerular injury in humans remain to be definitively identified. It is known that mercury shows significant renal tubular toxicity, and it is possible that this induces the release of self-antigens and resulting cytokine-associated inflammatory response [77,78];
- 6.
- In animal studies of mercury-induced autoimmunity, it has been shown that mercury exposure can clearly induce systemic autoimmunity in different animal species. This gives support to the biological plausibility of mercury as a factor in autoimmune diseases in humans. Many studies, mostly with mice, have demonstrated that mercury-induced autoimmunity can result from different modes of exposure, including oral ingestion of HgCl2, inhalation of mercury vapor, dental or periodontal implants containing dental amalgam, or subcutaneous injection [62,79,80,81,82,83].
- 7.
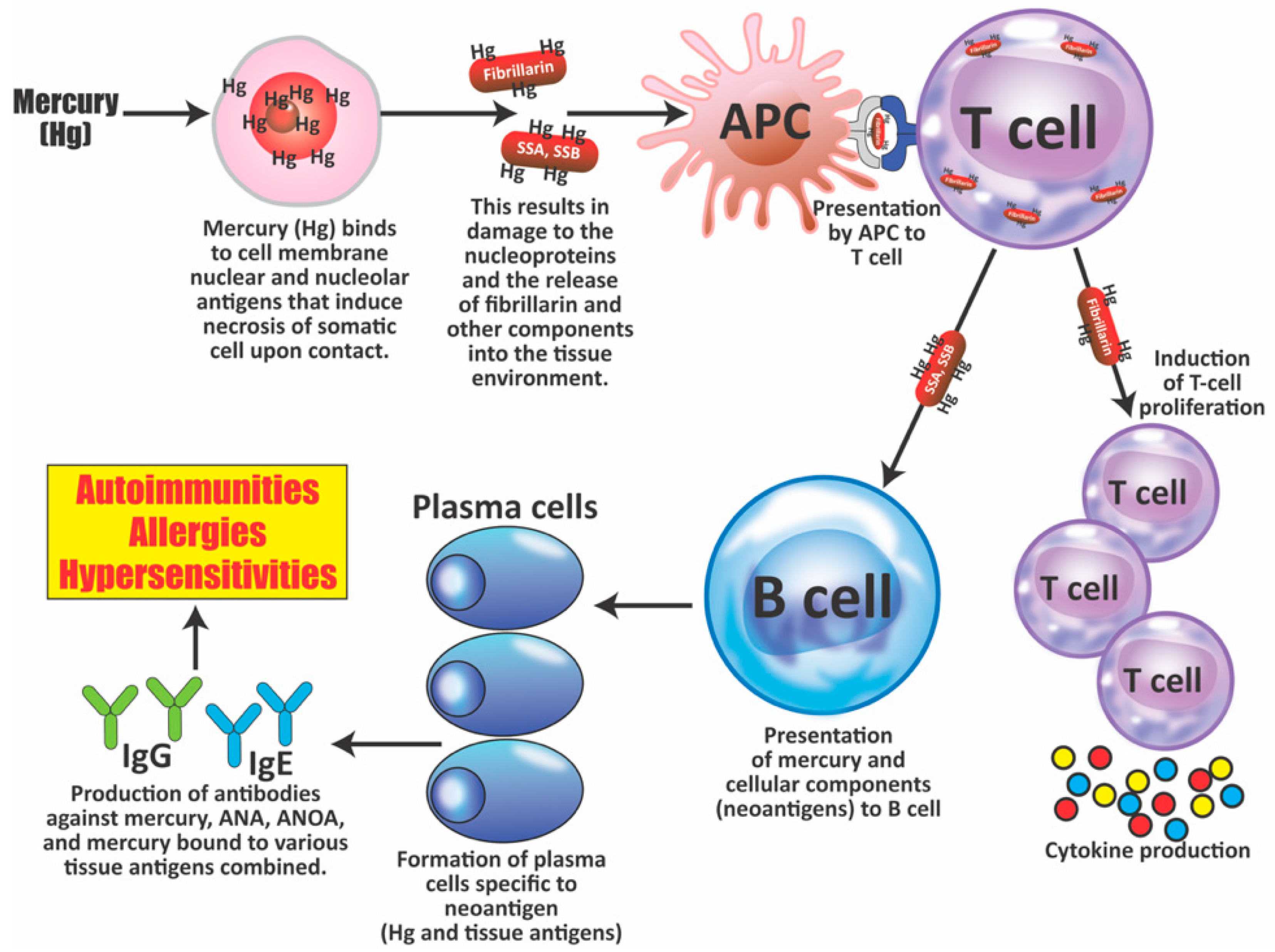
- Mercury-induced cell death can lead to the production of antibodies against the destroyed cell’s components. Antiglomerular basement membrane (GBM) autoantibodies have been reported in rats and rabbits; ANA has been found in rats. Mercury-induced ANA in mice was found to include ANoA, which has now been identified as an antibody against fibrillarin, a protein component of box C/D small nucleolar ribonucleoproteins (snoRNPs) particles, the main functions of which are methylation and the processing of pre-rRNA [84,85,86,87,88,89].
- 8.
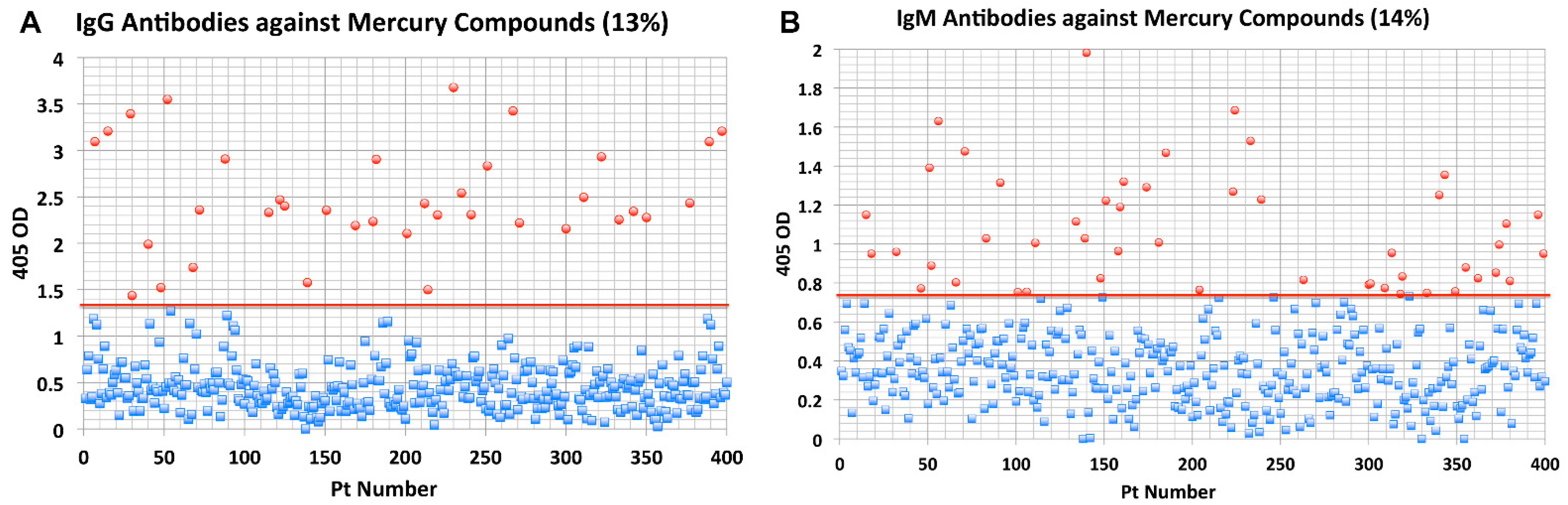
- Elevated levels of antibodies against xenobiotics (including mercury) have been found in a subgroup of healthy subjects. By acting as a hapten, mercury can bind to a high-molecular-weight carrier protein, such as human serum albumin (HSA), causing the immune system to mistakenly “recognize” self-tissue as an invader and launch an immune response against it, leading to autoimmunity. In one of our studies [30], we measured IgG and IgM antibodies against mercury and 11 other chemicals bound to HSA in the blood of supposedly healthy donors using ELISA methodology. We found that 13% (IgG) and 14% (IgM) of tested individuals showed significant antibody elevation against mercury (see Figure 6). The percentage of elevation against the other 11 chemicals ranged from 8% to 22% for IgG, and 13% to 18% for IgM.
4. Food Coloring and Autoimmunity
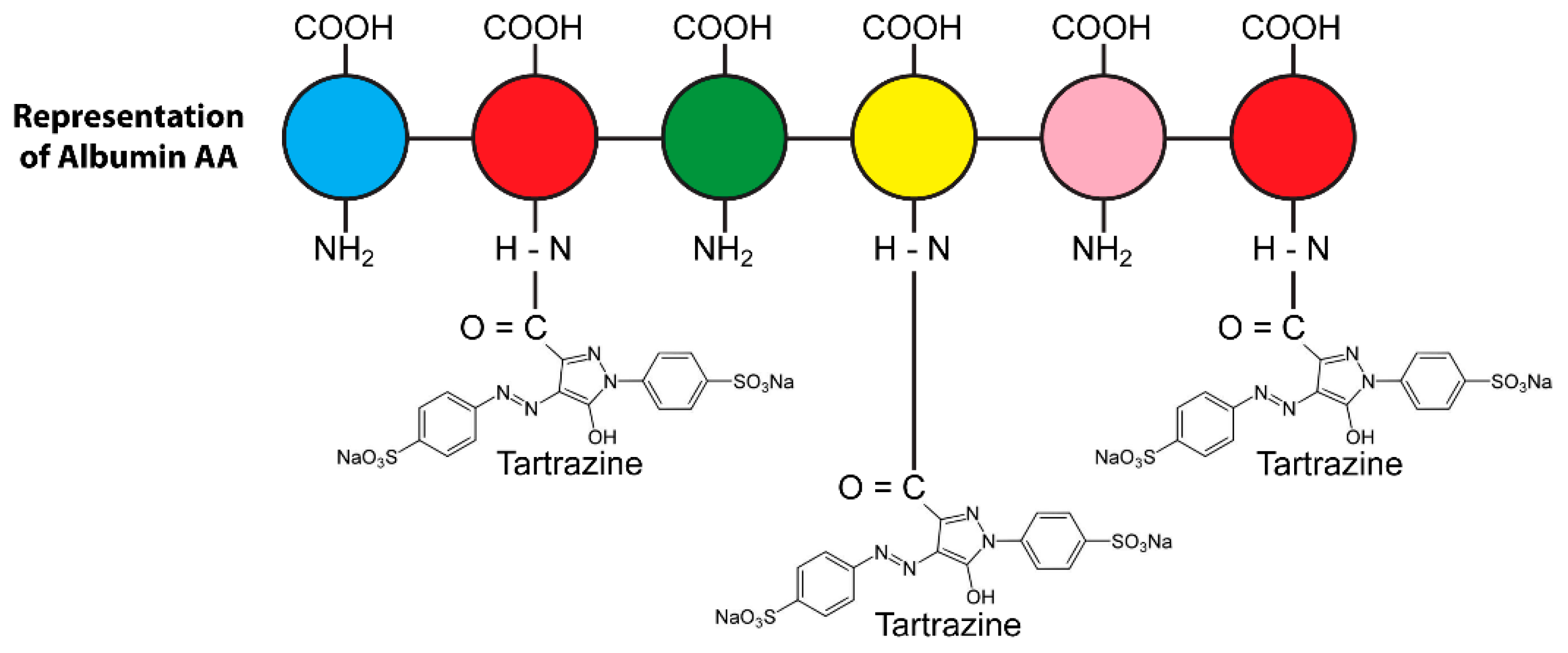
4.1. The Binding of Food Colors to Human Tissue Proteins Contributes to the Pathophysiology of Autoimmunity
4.2. The Effects of Food Coloring on Protein Digestibility
- Breakdown in oral tolerance;
- Decreased efficiency of digestive enzymes;
- Increase in intestinal permeability;
- Liver toxicity;
- Mitochondrial dysfunction;
- Hypersensitivity;
- Food immune reactivity;
- Asthma, allergic rhinitis, angioedema;
- Atopic dermatitis;
- Interference with neurotransmission;
- Neurobehavioral disorders;
- Reproductive abnormalities.
5. Reaction of the Immune System to Food Antigens and Its Contribution to Autoimmune Disorders
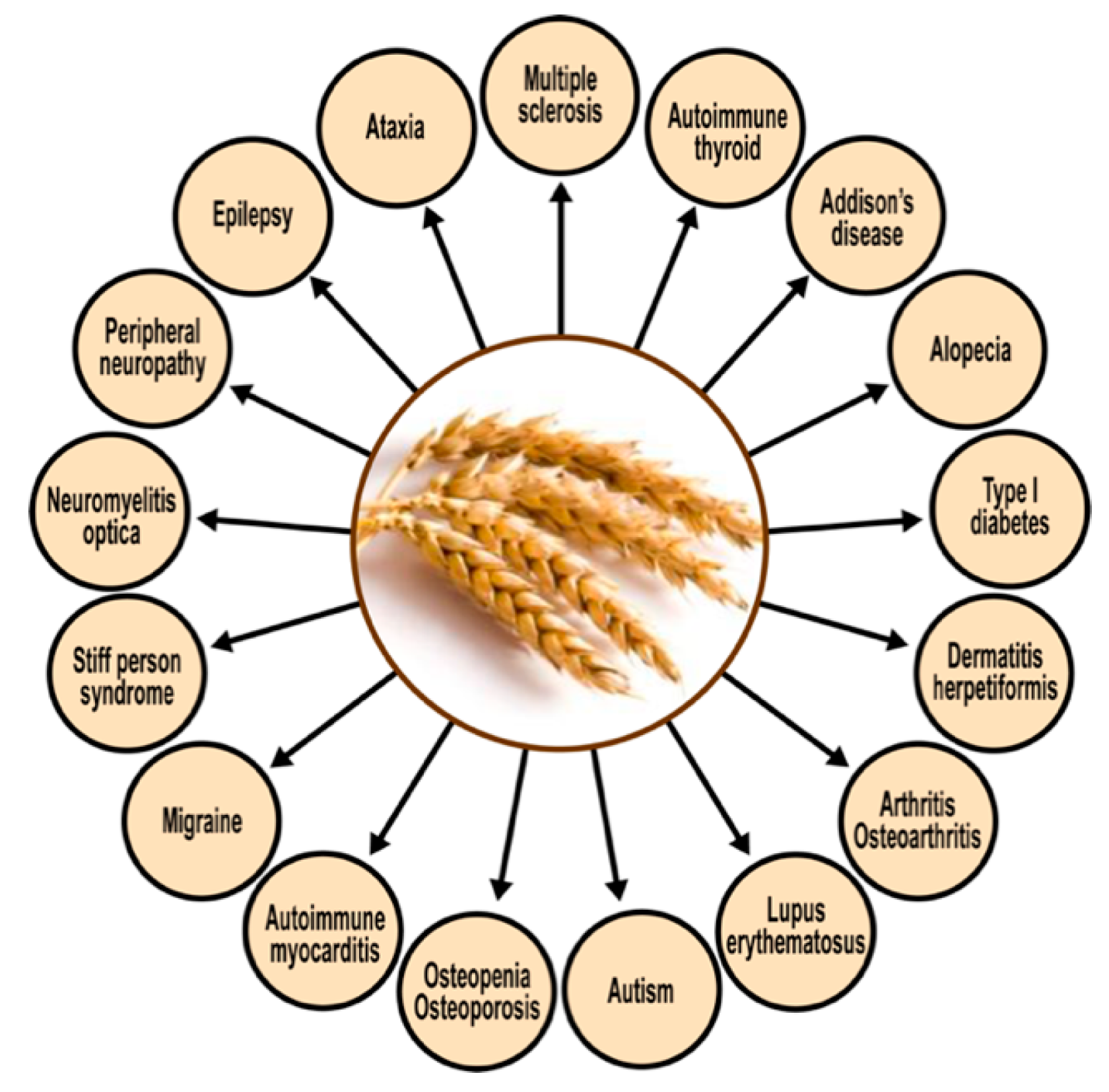
5.1. Autoimmunity, Wheat, and Milk
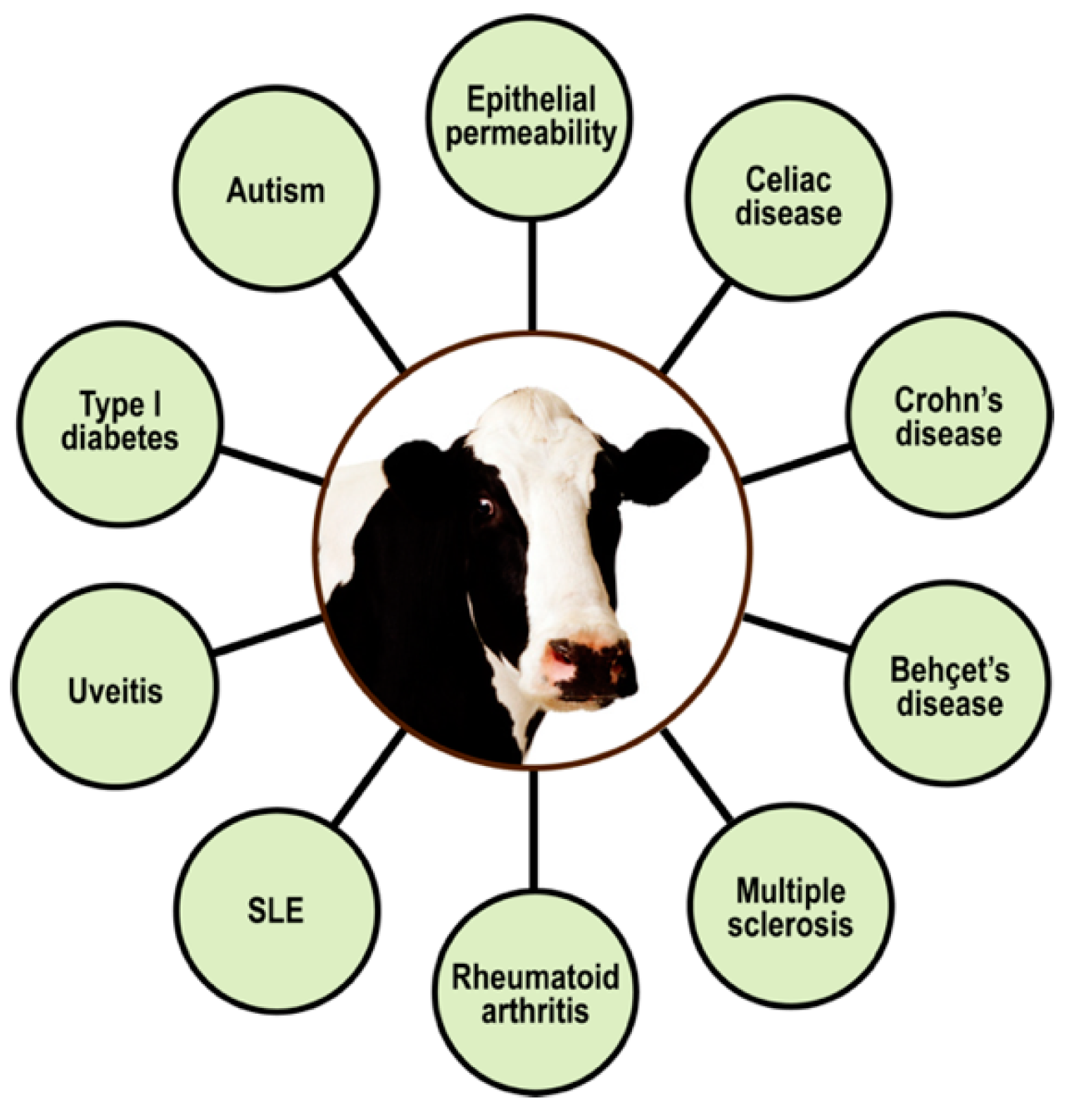
5.2. Immune Reactivity, Autoimmunity, and Milk Proteins
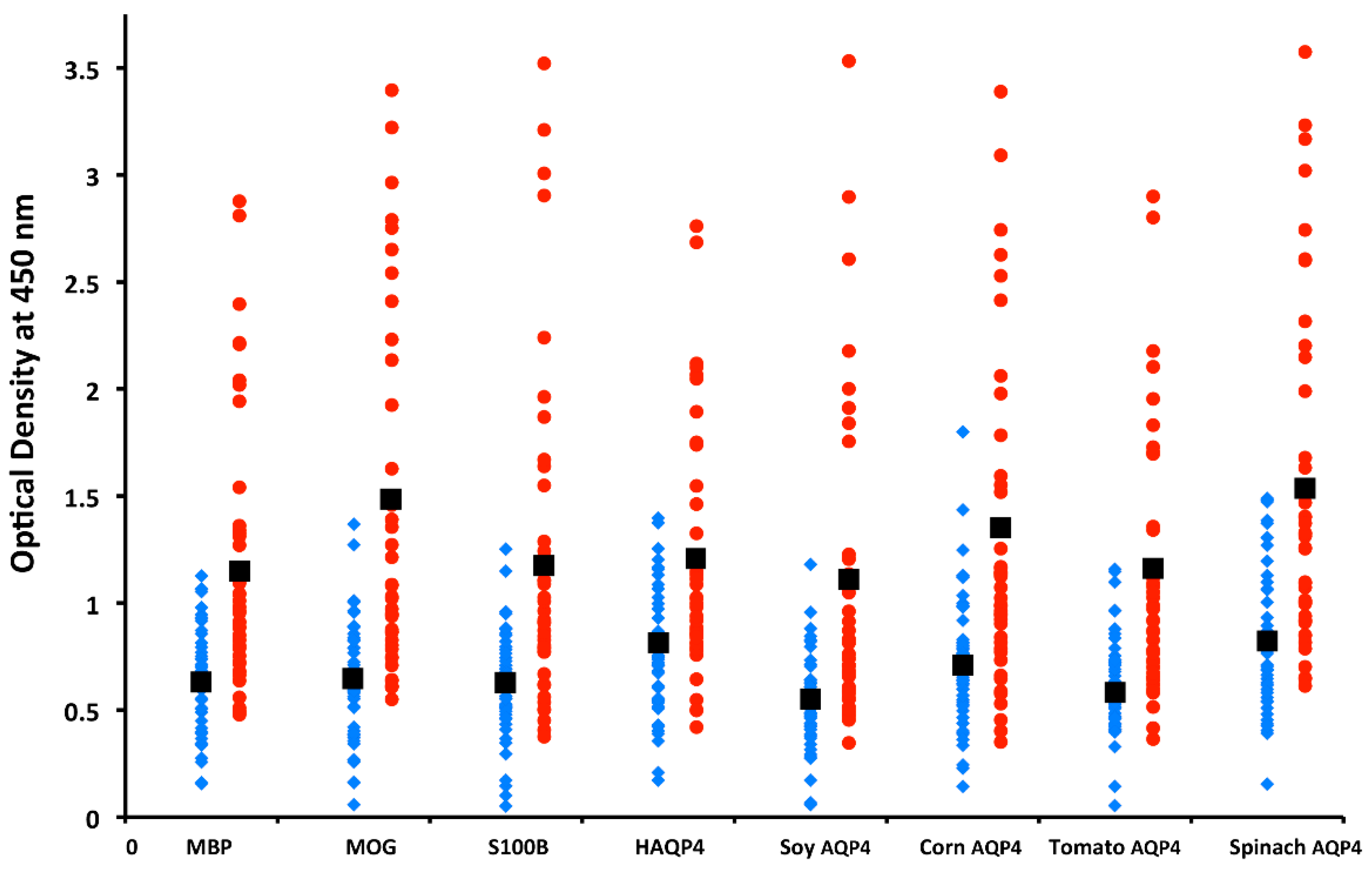
5.3. Neuroautoimmunity Due to Food Containing Aquaporins
5.4. Cross-Reactivity and Sequence Homology between Food Products and Alpha-Synuclein
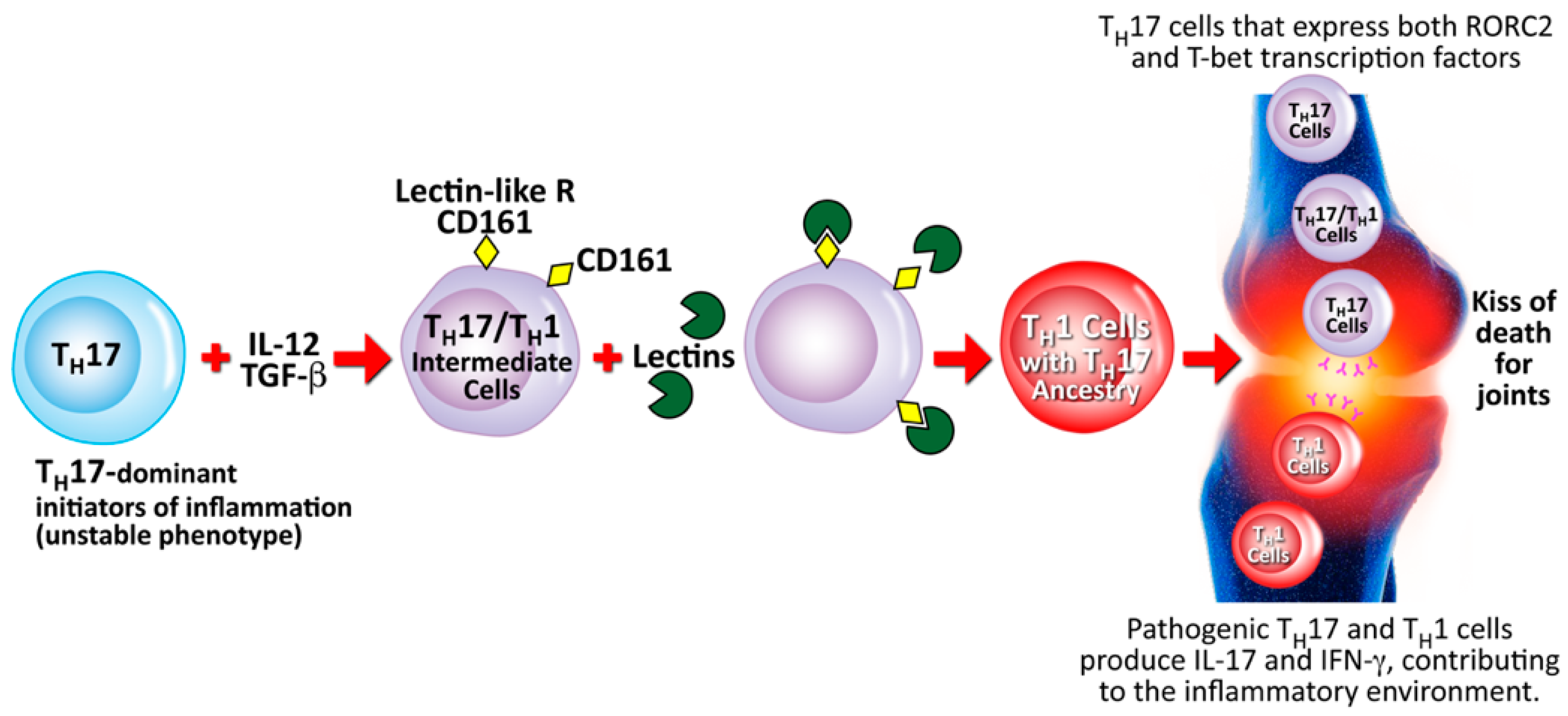
5.5. Contribution of Lectins and Agglutinins to Autoimmune Diseases
6. A Brief Look at Infections, Autoimmune Diseases, and the Hygiene Hypothesis
7. Conclusions
7.1. Strengths
7.2. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Opdenakker, G.; Abu El-Asrar, A.; Van Damme, J. Remnant epitopes generating autoimmunity: From model to useful paradigm. Trends Immunol. 2020, 41, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Wang, Q.; Korner, H.; Zhang, L.; Wei, W. Molecular mechanisms of T cells activation by dendritic cells in autoimmune diseases. Front. Pharmacol. 2018, 9, 642. [Google Scholar] [CrossRef]
- Steinman, R.M. Lasker basic medical research award. Dendritic cells versatile controllers of the immune system. Nat. Med. 2007, 13, 1155–1159. [Google Scholar] [CrossRef]
- Lebre, M.C.; Jongbloed, S.L.; Tas, S.W.; Smeets, T.J.; Mcinnes, I.B.; Tak, P.P. Rheumatoid arthritis synovium contains two subsets of CD83-DC-LAMP-dendritic cells with distinct cytokine profiles. Am. J. Pathol. 2008, 172, 940–950. [Google Scholar] [CrossRef]
- Tournadre, A.; Leniet, V.; Eljaafari, A.; Miossec, P. Immature muscle precursors are a source of interferon-beta in myositis: Role of Toll-like receptor 3 activation and contribution to HLA class I up-regulation. Arthritis Rheum. 2012, 64, 533–541. [Google Scholar] [CrossRef]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappucio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Min, S.Y.; Park, K.S.; Cho, M.L.; Kang, J.W.; Cho, Y.G.; Hwang, S.Y.; Park, M.J.; Yoon, C.H.; Min, J.K.; Lee, S.H.; et al. Antigen-induced, tolerogenic CD11c+,CD11b+ dendritic cells are abundant in Peyer’s patches during the induction of oral tolerance to type II collagen and suppress experimental collagen-induced arthritis. Arthritis Rheum. 2006, 54, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, G.X.; Chen, Y.; Xu, H.; Fitzgerald, D.C.; Zhao, Z.; Rostami, A. CD11c+CD11b+ dendritic cells play an important role in intravenous tolerance and the suppression of experimental autoimmune encephalomyelitis. J. Immunol. 2008, 181, 2483–2493. [Google Scholar] [CrossRef]
- Audiger, C.; Rahman, M.J.; Yun, T.J.; Tarbell, K.V.; Lesage, S. The importance of dendritic cells in maintaining immune tolerance. J. Immunol. 2017, 198, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.T.; Chang, C.; Gershwin, M.E.; Lian, Z.X. Development of autoantibodies precedes clinical manifestations of autoimmune diseases: A comprehensive review. J. Autoimmun. 2017, 83, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Notkins, A.L. New predictors of disease. Sci. Am. 2007, 296, 72–79. [Google Scholar] [CrossRef]
- Autoimmune Diseases and Your Environment. Available online: https://www.niehs.nih.gov/health/materials/autoimmune_diseases_and_your_environment_508.pdf (accessed on 2 November 2021).
- Dinse, G.E.; Parks, C.G.; Weinberg, C.R.; Co, C.A.; Wilkerson, J.; Zeldin, D.C.; Chan, E.K.L.; Miller, F.W. Increasing prevalence of antinuclear antibodies in the United States. Arthritis Rheumatol. 2020, 72, 1026–1035. [Google Scholar] [CrossRef]
- Autoimmune Disease List. Available online: https://www.autoimmuneinstitute.org/resources/autoimmune-disease-list/ (accessed on 2 November 2021).
- Vojdani, A. A potential link between environmental triggers and autoimmunity. Autoimmune Dis. 2014, 2014, 437231. [Google Scholar] [CrossRef]
- Schmidt, C.W. Questions persist. Environmental factors in autoimmune disease. Environ. Health Perspect. 2011, 119, A249–A253. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Ahn, B.S.; Cha, D.; Jang, W.W.; Choi, E.; Park, S.; Park, J.H.; Oh, J.; Jung, D.E.; Park, H.; et al. Understanding the immunopathogenesis of autoimmune diseases by animal studies using gene modulation: A comprehensive review. Autoimmun. Rev. 2020, 19, 102469. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef]
- Rappaport, S.M.; Smith, M.T. Epidemiology, environment and disease risks. Science 2010, 330, 460–461. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, R.; Scymanski, E.L.; Barabasi, A.L.; Miller, G.W. The exposome and health: Where chemistry meets biology. Science 2020, 367, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Robinson, O.; Chadeau-Hyam, M.; Dehghan, A.; Mudway, I.; Dagnino, S. What is new in the exposome? Environ. Int. 2020, 143, 105887. [Google Scholar] [CrossRef]
- Leffers, H.C.B.; Lange, T.; Collins, C.; Ulff-Møller, C.J.; Jacobsen, S. The study of interactions between genome and exposome in the development of systemic lupus erythematosus. Autoimmun. Rev. 2019, 18, 382–392. [Google Scholar] [CrossRef]
- Vojdani, A.; O’Bryan, T.; Green, J.A.; McCandless, J.; Woeller, K.N.; Vojdani, E.; Nourian, A.A. Immune response to dietary proteins, gliadin and cerebellar peptides in children with autism. Nutr. Neurosci. 2004, 7, 151–161. [Google Scholar] [CrossRef]
- Vojdani, A.; Kharrazian, D.; Mukherjee, P.S. The prevalence of antibodies against wheat and milk proteins in blood donors and their contribution to neuroimmune reactivities. Nutrients 2014, 6, 15–36. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Tarash, I. Cross-reaction between gliadin and different food and tissue antigens. Food Nutr. Sci. 2013, 44, 20–32. [Google Scholar] [CrossRef]
- Vojdani, A. Lectins, agglutinins, and their role in autoimmune reactivities. Altern. Ther. Health Med. 2015, 21, 46–51. [Google Scholar]
- Vojdani, A.; Afar, D.; Vojdani, E. Reaction of lectin-specific antibody with human tissue: Possible contributions to autoimmunity. J. Immunol. Res. 2020, 2020, 1438957. [Google Scholar] [CrossRef]
- Vojdani, A. Reaction of anti-food antibodies with different tissue antigens. Int. J. Food Sci. Technol. 2020, 55, 1800–1815. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E. Immunoreactivity of anti-AbP-42 specific antibody with toxic chemicals and food antigens. J. Alzheimers Dis. Parkinsonism 2018, 8, 441. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E. Food-Associated Autoimmunities: When Food Breaks Your Immune System, 1st ed.; A&G Press: Los Angeles, CA, USA, 2019. [Google Scholar]
- Vojdani, A.; Kharrazian, D.; Mukherjee, P.S. Elevated levels of antibodies against xenobiotics in a subgroup of healthy subjects. Nutrients 2015, 35, 383–397. [Google Scholar]
- Vojdani, A.; Gushgari, L.R.; Vojdani, E. Interaction between food antigens and the immune system: Association with autoimmune disorders. Autoimmun. Rev. 2020, 19, 102459. [Google Scholar] [CrossRef]
- Lerner, A.; Matthias, T. Changes in intestinal tight junction permeability associated with industrial food additives explain the rising incidence of autoimmune disease. Autoimmun. Rev. 2015, 14, 479–489. [Google Scholar] [CrossRef]
- Li, B.; Selmi, C.; Tang, R.; Gershwin, M.; Ma, X. The microbe and autoimmunity: A paradigm from the gut-liver axis. Mol. Immunol. 2018, 15, 1–15. [Google Scholar]
- Opazo, M.; Ortega-Rocha, E.M.; Coronado-Arrazola, I.; Bonifaz, L.C.; Boudin, H.; Neunlist, M.; Bueno, S.M.; Kalergis, A.M.; Riedel, C.A. Intestinal microbiota influences non-intestinal related autoimmune diseases. Front. Microbiol. 2018, 9, 432. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, S.M.; Knip, M. Nutritional risk predictors of β cell autoimmunity and type 1 diabetes at a young age. Am. J. Clin. Nutr. 2003, 78, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Coucke, F. Food intolerance in patients with manifest autoimmunity. Observational study. Autoimmun. Rev. 2018, 17, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Parkkinen, J. Aberrant lectin-binding activity of immunoglobulin G in serum from rheumatoid arthritis patients. Clin. Chem. 1989, 35, 1638–1643. [Google Scholar] [CrossRef]
- Hvatum, M.; Kanerud, L.; Hällgren, R.; Brandtzaeg, P. The gut-joint axis: Cross reactive food antibodies in rheumatoid arthritis. Gut 2006, 55, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Conigliaro, P.; Triggianese, P.; De Martino, E.; Fonti, G.L.; Chimenti, M.S.; Sunzini, F.; Viola, A.; Canofari, C.; Perricone, R. Challenges in the treatment of rheumatoid arthritis. Autoimmun. Rev. 2019, 18, 706–713. [Google Scholar] [CrossRef]
- Nielsen, S.M.; Zobbe, K.; Kristensen, L.E.; Christensen, R. Nutritional recommendations for gout: An update from clinical epidemiology. Autoimmun. Rev. 2018, 17, 1090–1096. [Google Scholar] [CrossRef]
- Holzbauer, S.M.; DeVries, A.S.; Sejvar, J.J.; Lees, C.H.; Adjemian, J.; McQuiston, J.H.; Medus, C.; Lexau, C.A.; Harris, J.R.; Recuenco, S.E.; et al. Epidemiologic investigation of immune-mediated polyradiculoneuropathy among abattoir workers exposed to porcine brain. PLoS ONE 2010, 5, e9782. [Google Scholar] [CrossRef]
- Gershteyn, I.M.; Ferreira, L.M.R. Immunodietica: A data-driven approach to investigate interactions between diet and autoimmune disorders. J. Transl. Autoimmun. 2019, 1, 100003. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.N.; Xu, Z.; Wu, G.C.; Mao, Y.M.; Liu, L.N.; Qian, W.; Dan, Y.L.; Tao, S.S.; Zhang, Q.; Sam, N.B.; et al. Emerging role of air pollution in autoimmune diseases. Autoimmun. Rev. 2019, 18, 607–614. [Google Scholar] [CrossRef]
- Selmi, C.; Lu, Q.; Humble, M.C. Heritability versus the role of the environment in autoimmunity. J. Autoimmun. 2012, 39, 249–252. [Google Scholar] [CrossRef]
- Pollard, K.M.; Hultman, P.; Kono, D.H. Toxicology of autoimmune diseases. Chem. Res. Toxicol. 2010, 23, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Pollard, K.M.; Campbell, A.W. Environmental triggers and autoimmunity. Autoimmune Dis. 2014, 2014, 798029. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.F.; Wang, G. Environmental agents, oxidative stress and autoimmunity. Curr. Opin. Toxicol. 2016, 7, 22–27. [Google Scholar] [CrossRef]
- Khan, M.F.; Wang, H. Environmental exposure and autoimmune diseases: Contribution to gut microbiome. Front. Immunol. 2019, 10, 3094. [Google Scholar] [CrossRef]
- Suzuki, T.; Hidaka, T.; Kumagai, Y.; Yamamoto, M. Environmental pollutants and the immune response. Nat. Immunol. 2020, 21, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Bigazzi, P.E. Autoimmunity caused by xenobiotics. Toxicology 1997, 119, 1–21. [Google Scholar] [CrossRef]
- Selmi, C. Mechanisms of environmental influences on human autoimmunity: A National Institute of environmental health sciences expert panel workshop. J. Autoimmun. 2012, 39, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Barragan-Martinez, C.; Speck-Hernandez, C.A.; Montoya-Ortiz, G. Organic solvents as risk factor for autoimmune diseases: A systemic review and meta-analysis. PLoS ONE 2012, 7, 51506. [Google Scholar] [CrossRef]
- Baccarelli, A.; Wright, R.O.; Bollati, V. Rapid DNA methylation changes after exposure to traffic particles. Am. J. Respir. Crit. Care Med. 2009, 179, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.M.; Cauvi, D.M.; Mayeux, J.M.; Toomey, C.B.; Peiss, A.K.; Hultman, P.; Kono, D.H. Mechanisms of environment-induced autoimmunity. Ann. Rev. Pharmacol. Toxicol. 2021, 61, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.M.; Christy, J.M.; Cauvi, D.M.; Kono, D.H. Environmental xenobiotics exposure and autoimmunity. Curr. Opin. Toxicol. 2018, 10, 15–22. [Google Scholar] [CrossRef]
- Miller, F.W.; Alfredsson, L.; Costenbader, K.H.; Kamen, D.L.; Nelson, L.M.; Norris, J.M.; De Roos, A.J. Epidemiology of environmental exposures and human autoimmune diseases: Findings from a National Institute of environmental health sciences expert panel workshop. J. Autoimmun. 2012, 39, 259–271. [Google Scholar] [CrossRef]
- Pollard, K.M. Gender differences in autoimmunity associated with exposure to environmental factors. J. Autoimmun. 2012, 38, J177–J186. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Kono, D.H.; Bacczala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.M.; Kono, D.H. Requirements for innate immune pathways in environmentally induced autoimmunity. BMC Med. 2013, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Kono, D.H.; Bacczala, R.; Theofilopoulos, A.N. TLRs and interferons: A central paradigm in autoimmunity. Curr. Opin. Immunol. 2013, 25, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.T.; Scatizzi, J.C.; Gahan, J.D.; Lawson, B.R.; Baccala, R.; Pollard, K.M.; Beutler, B.A.; Theofilopoulos, A.N.; Kono, D.H. Role of nucleic acid-sensing TLRs in diverse autoantibody specificities and anti-nuclear antibody-producing B cells. J. Immunol. 2013, 190, 4982–4990. [Google Scholar] [CrossRef]
- Schiraldi, M.; Monetier, M. How can a chemical element elicit complex immunopathology? Lessons from mercury-induced autoimmunity. Trends Immunol. 2009, 30, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Peana, M.; Dadar, M.; Chirumbolo, S.; Aaseth, J.; Martins, N. Mercury-induced autoimmunity: Drifting from micro to macro concerns on autoimmune disorder. Clin. Immunol. 2020, 213, 108352. [Google Scholar] [CrossRef]
- Nordlind, K. Fractionation of human thymocytes and peripheral blood lymphocytes on Percoll density gradients and DNA synthesis stimulating effect of mercuric chloride. Int. Arch. Allergy Appl. Immunol. 1984, 75, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Hebel, S.; Engelhardt, G.; Rink, L. Ethylmercury and Hg2+ induce the formation of neutrophil extracellular traps (NETs) by human neutrophil granulocytes. Arch. Toxicol. 2016, 90, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.M.; Nyland, J.F.; Silva, I.A.; Ventura, A.M.; de Souza, J.M.; Silbergeld, E.K. Mercury exposure, serum antinuclear/antinucleolar antibodies, and serum cytokine levels in mining populations in Amazonian Brazil: A cross-sectional study. Environ. Res. 2010, 110, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Nyland, J.F.; Fillion, M.; Barbosa, F., Jr.; Shirley, D.L.; Chine, C.; Lemire, M.; Mergler, D.; Silbergeld, E.K. Biomarkers of methylmercury exposure immunotoxicity among fish consumers in Amazonian Brazil. Environ. Health. Perspect. 2011, 119, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.; Erdei, E.; Rubin, R.L.; Miller, C.; Ducheneaux, C.; O’Leary, M.; Pacheco, B.; Mahler, M.; Henderson, P.N.; Pollard, K.M.; et al. Mercury, autoimmunity, and environmental factors on Cheyenne River Sioux tribal lands. Autoimmune Dis. 2014, 2014, 325461. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.M.; Meliker, J.R. Mercury and thyroid autoantibodies in U.S. women, NHANES 2007–2008. Environ. Int. 2012, 40, 39–43. [Google Scholar] [CrossRef]
- Somers, E.C.; Ganser, M.A.; Warren, J.S.; Basu, N.; Wang, L.; Zick, S.M.; Park, S.K. Mercury exposure and antinuclear antibodies among females of reproductive age in the United States: NHANES. Environ. Health Perspect. 2015, 123, 792–798. [Google Scholar] [CrossRef]
- Miller, S.; Pallan, S.; Gangji, A.S.; Lukic, D.; Clase, C.M. Mercury-associated nephrotic syndrome: A case report and systematic review of the literature. Am. J. Kidney Dis. 2013, 62, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.Y. Inorganic mercury poisoning associated with skin-lightening cosmetic products. Clin. Toxicol. 2011, 49, 886–891. [Google Scholar] [CrossRef]
- Li, S.J.; Zhang, S.H.; Chen, H.P.; Zeng, C.H.; Zheng, C.X.; Li, L.S.; Liu, Z.H. Mercury-induced membranous nephropathy: Clinical and pathological features. Clin. J. Am. Soc. Nephrol. 2010, 5, 439–444. [Google Scholar] [CrossRef]
- Harrison, D.J.; Thomson, D.; MacDonald, M.K. Membranous glomerulonephritis. J. Clin. Pathol. 1986, 39, 167–171. [Google Scholar] [CrossRef]
- Tang, H.L.; Chu, K.H.; Mak, Y.F.; Lee, W.; Cheuk, A.; Yim, K.F.; Fung, K.S.; Chan, H.W.; Tong, K.L. Minimal change disease following exposure to mercury-containing skin lightening cream. Hong Kong Med. J. 2006, 12, 316–318. [Google Scholar]
- Orr, S.E.; Bridges, C.C. Chronic kidney disease and exposure to nephrotoxic metals. Int. J. Mol. Sci. 2017, 18, 1039. [Google Scholar] [CrossRef] [PubMed]
- Wagrowska-Danilewicz, M.; Danilewicz, M.; Zbrog, Z. Mercury-induced nephrotic syndrome: A case report and review of the literature. Pol. J. Pathol. 2014, 65, 322–326. [Google Scholar] [CrossRef]
- Whitekus, M.J.; Santini, R.P.; Rosenspire, A.J.; McCabe, M.J., Jr. Protection against CD95-mediated apoptosis by inorganic mercury in Jurkat T cells. J. Immunol. 1999, 162, 7162–7170. [Google Scholar]
- McCabe, M.J., Jr.; Whitekus, M.J.; Hyun, J.; Eckles, K.G.; McCollum, G.; Rosenspire, A.J. Inorganic mercury attenuates CD95-mediated apoptosis by interfering with formation of the death inducing signaling complex. Toxicol. Appl. Pharmacol. 2003, 190, 146–156. [Google Scholar] [CrossRef]
- Ziemba, S.E.; McCabe, M.J., Jr.; Rosenspire, A.J. Inorganic mercury dissociates preassembled Fas/CD95 receptor oligomers in T lymphocytes. Toxicol. Appl. Pharmacol. 2005, 206, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Mattingly, R.R.; Felczak, A.; Chen, C.C.; McCabe, M.J., Jr.; Rosenspire, A.J. Low concentrations of inorganic mercury inhibit Ras activation during T cell receptor-mediated signal transduction. Toxicol. Appl. Pharmacol. 2001, 176, 162–168. [Google Scholar] [CrossRef]
- Ziemba, S.E.; Mattingly, R.R.; McCabe, M.J., Jr.; Rosenspire, A.J. Inorganic mercury inhibits the activation of LAT in T-cell receptor-mediated signal transduction. Toxicol. Sci. 2006, 89, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Sapin, C.; Druet, E.; Druet, P. Induction of anti-glomerular basement membrane antibodies in the Brown-Norway rat by mercuric chloride. Clin. Exp. Immunol. 1977, 28, 173–179. [Google Scholar]
- Roman-Franco, A.A.; Turiello, M.; Albini, B.; Ossi, E.; Milgrom, F.; Andres, G.A. Anti-basement membrane antibodies and antigen—Antibody complexes in rabbits injected with mercuric chloride. Clin. Immunol. Immunopathol. 1978, 9, 464–481. [Google Scholar] [CrossRef]
- Weening, J.J.; Fleuren, G.J.; Hoedemaeker, P.J. Demonstration of antinuclear antibodies in mercuric chloride-induced glomerulopathy in the rat. Lab. Investig. 1978, 39, 405–411. [Google Scholar] [PubMed]
- Robinson, C.J.; Abraham, A.A.; Balazs, T. Induction of anti-nuclear antibodies by mercuric chloride in mice. Clin. Exp. Immunol. 1984, 58, 300–306. [Google Scholar]
- Hultman, P.; Enestrom, S.; Pollard, K.M.; Tan, E.M. Anti-fibrillarin autoantibodies in mercury-treated mice. Clin. Exp. Immunol. 1989, 78, 470–477. [Google Scholar] [PubMed]
- Reuter, R.; Tessars, G.; Vohr, H.W.; Gleichmann, E.; Luhrmann, R. Mercuric chloride induces autoantibodies against U3 small nuclear ribonucleoprotein in susceptible mice. Proc. Natl. Acad. Sci. USA 1989, 86, 237–241. [Google Scholar] [CrossRef]
- Hultman, P.; Bell, L.J.; Enestrom, S.; Pollard, K.M. Murine susceptibility to mercury. I. Autoantibody profiles and systemic immune deposits in inbred, congenic, and intra-H-2 recombinant strains. Clin. Immunol. Immunopathol. 1992, 65, 98–109. [Google Scholar] [CrossRef]
- Takeuchi, K.; Turley, S.J.; Tan, E.M.; Pollard, K.M. Analysis of the autoantibody response to fibrillarin in human disease and murine models of autoimmunity. J. Immunol. 1995, 154, 961–971. [Google Scholar] [PubMed]
- Pollard, K.M.; Lee, D.K.; Casiano, C.A.; Bluthner, M.; Johnston, M.M.; Tan, E.M. The autoimmunity-inducing xenobiotic mercury interacts with the autoantigen fibrillarin and modifies its molecular and antigenic properties. J. Immunol. 1997, 158, 3521–3528. [Google Scholar] [PubMed]
- Pollard, K.M.; Pearson, D.L.; Bluthner, M.; Tan, E.M. Proteolytic cleavage of a self-antigen following xenobiotic-induced cell death produces a fragment with novel immunogenic properties. J. Immunol. 2000, 165, 2263–2270. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, C. Immune reactivity to food coloring. Altern. Ther. Health Med. 2015, 21, 52–62. [Google Scholar] [PubMed]
- Carter, C.M.; Urbanowicz, M.; Hemsley, R.; Mantilla, L.; Strobel, S.; Graham, P.J.; Taylor, E. Effects of a few food diet in attention deficit disorder. Arch. Dis. Child. 1993, 21, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Boris, M.; Mandel, F.S. Foods and additives are common causes of the attention deficit hyperactive disorder in children. Ann. Allergy 1994, 72, 462–468. [Google Scholar] [PubMed]
- Lucova, M.; Hojerova, J.; Pazourekova, S.; Klimova, Z. Absorption of triphenylmethane dyes Brilliant Blue and Patent Bliue through intact skin, shaven skin and lingual mucosa from daily life products. Food Chem. Toxicol. 2013, 52, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.P.; Raymundo, A.; Sousa, I.; Empis, J.; Franco, J.M. Colored food emulsions—Implications of pigment addition on the rheological behavior and microstructure. Food Biophys. 2006, 1, 216–227. [Google Scholar] [CrossRef]
- European Food Safety Authority Panel on Food Additives and Nutrient Sources. Scientific opinion and the re-evaluation tartrazine (E 102). Eur. Food Saf. Auth. 2009, 7, 1331–1382. [Google Scholar]
- Abdullah, S.; Badaruddin, M.; Sayeed, S.A.; Ali, R.; Riaz, M.N. Binding ability of allura red with food proteins and its impact on protein digestibility. Food Chem. 2008, 110, 605–610. [Google Scholar] [CrossRef]
- Badaruddin, M.; Abdullah, S.U.; Sayeed, S.A.; Ali, R.; Riaz, M.N. Sunset yellow: A food for protein staining with SDS-PAGE. Cereal Food World 2007, 52, 12–14. [Google Scholar] [CrossRef]
- Saeed, S.M.; Abdullah, S.U.; Sayeed, S.A.; Ali, R. Food protein: Food color interactions and its application in rapid protein assay. Czech. J. Food. Sci. 2010, 28, 506–513. [Google Scholar] [CrossRef]
- Weliky, N.; Heiner, D.C. Hypersensitivity to chemicals: Correlation of tartrazine hypersensitivity with characteristic serum IgD and IgE immune response patterns. Clin. Allergy 1980, 10, 375–395. [Google Scholar] [CrossRef]
- Saeed, S.M.; Sayeed, S.A.; Ashraf, S.; Nassimunisa; Batool, F.; Ali, R.; Naz, S.; Siddiqi, R. Investigations of in vitro digestibility of proteins bound to food colors. J. Pharm. Nutr. Sci. 2011, 1, 34–40. [Google Scholar] [CrossRef]
- Katrahalli, U.; Kalanur, S.S.; Seetharamappa, J. Interaction of bioactive coomassie Brilliant Blue G with protein: Insights from spectroscopic methods. Sci. Pharm. 2010, 78, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Mathavan, V.M.; Boh, B.K.; Tayyab, S. Characterization of erythrosine B binding to bovine serum albumin and bilirubin displacement. Indian J. Biochem. Biophys. 2009, 46, 325–331. [Google Scholar]
- Li, Y.; Wei, H.; Liu, R. A probe to study the toxic interaction of tartrazine with bovine hemoglobin at the molecular level. Luminescence 2014, 29, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Guendouz, M.; Mehedi, N.; Zaoui, C.; Saidi, D.; Khéroua, O. Immune response after tartrazine subchronic ingestion in Swiss albino mice. Int. J. Pharm. Pharm. Sci. 2013, 5, 584–592. [Google Scholar]
- Vojdani, A. For the assessment of intestinal permeability, size matters. Altern. Ther. Health Med. 2013, 19, 12–24. [Google Scholar] [PubMed]
- Zhang, G.; Ma, Y. Mechanistic and conformational studies on the interaction of food dye amaranth with human serum albumin by multispectroscopic methods. Food Chem. 2013, 136, 442–449. [Google Scholar] [CrossRef]
- Schrander, J.J.; Marcelis, C.; de Vries, M.P.; van Santen-Hoeufft, H.M. Does food intolerance play a role in juvenile chronic arthritis? Br. J. Rheumatol. 1997, 36, 905–908. [Google Scholar] [CrossRef]
- Molberg, Ø.; Sollid, L.M. A gut feeling for joint inflammation—Using celiac disease to understand rheumatoid arthritis. Trends Immunol. 2006, 27, 188–194. [Google Scholar] [CrossRef]
- Lunardi, C.; Nanni, L.; Tiso, M.; Mingari, M.C.; Bason, C.; Oliveri, M.; Keller, B.; Millo, R.; De Sandre, G.; Corrocher, R.; et al. Glycine-rich cell wall proteins act as specific antigen targets in autoimmune and food allergic disorders. Int. Immunol. 2000, 12, 647–657. [Google Scholar] [CrossRef]
- Baboonian, C.; Halliday, D.; Venables, P.J.; Pawlowski, T.; Millman, G.; Maini, R.N. Antibodies in rheumatoid arthritis react specifically with the glycine alanine repeat sequence of Epstein-Barr nuclear antigen-1. Rheumatol. Int. 1989, 9, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Baboonian, C.; Venables, P.J.; Williams, D.G.; Williams, R.O.; Maini, R.N. Cross reaction of antibodies to glycine/alanine repeat sequence of Epstein-Barr virus nuclear antigen-1 with collagen, cytokeratin, and actin. Ann. Rheum. Dis. 1991, 50, 772–775. [Google Scholar] [CrossRef]
- Ostenstad, B.; Dybwad, A.; Lea, T.; Forre, O.; Vinje, O.; Sioud, M. Evidence for monoclonal expansion of synovial T cells bearing V alpha 2.1/V beta 5.5 gene segments and recognizing a synthetic peptide that shares homology with a number of putative autoantigens. Immunology 1995, 86, 168–175. [Google Scholar]
- Zuidmeer, L.; Goldhahn, K.; Rona, R.J.; Gislason, D.; Madsen, C.; Summers, C.; Sodergen, E.; Dahlstrom, J.; Lindner, T.; Sigurdardottir, S.T.; et al. The prevalence of plant food allergies: A systematic review. J. Allergy Clin. Immunol. 2008, 121, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Lack, G. Epidemiologic risks for food allergy. J. Allergy Clin. Immunol. 2008, 121, 1331–1336. [Google Scholar] [CrossRef]
- Bousquet, J.; Bjὂrkstén, B.; Bruijnzeel-Koomen, C.A.; Huggett, A.; Ortolani, C.; Warner, J.O.; Smith, M. Scientific criteria and the selection of allergenic foods for product labeling. Allergy 1998, 53, 3–21. [Google Scholar] [CrossRef]
- Arentz-Hansen, H.; Kὂrner, R.; Molberg, Ø.; Quarsten, H.; Vader, W.; Kooy, Y.M.C.; Lundin, K.E.A.; Koning, F.; Roepstorff, P.; Sollid, L.M.; et al. The intestinal T cell response to alpha-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J. Exp. Med. 2000, 191, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Arentz-Hansen, H.; McAdam, S.N.; Molberg, Ø.; Fleckenstein, B.; Lundin, K.E.A.; Jørgensen, T.J.D.; Jung, G.; Roepstorff, P.; Sollid, L.M. Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology 2002, 123, 803–809. [Google Scholar] [CrossRef]
- Tollefsen, S.; Arentz-Hansen, H.; Fleckenstein, B.; Molberg, Ø.; Ráki, M.; Kwok, W.W.; Jung, G.; Lundin, K.E.A.; Sollid, L.M. HLA-DQ2 and -DQ8 signatures of gluten T cell epitopes in celiac disease. J. Clin. Investig. 2006, 116, 2226–2236. [Google Scholar] [CrossRef]
- Camarca, A.; Anderson, R.P.; Mamone, G.; Fierro, O.; Facchiano, A.; Costantini, S.; Zanzi, D.; Sidney, J.; Auricchio, S.; Sette, A.; et al. Intestinal T cell responses to gluten peptides are largely heterogeneous: Implications for a peptide-based therapy in celiac disease. J. Immunol. 2009, 182, 4158–4166. [Google Scholar] [CrossRef]
- Tye-Din, J.A.; Stewart, J.A.; Dromey, J.A.; Beissbarth, T.; van Heel, D.A.; Tatham, A.; Henderson, K.; Mannering, S.I.; Gianfrani, C.; Jewell, D.P.; et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci. Transl. Med. 2010, 2, 41ra51. [Google Scholar] [CrossRef] [PubMed]
- Sjὂstrὂm, H.; Lundin, K.E.; Molberg, Ø.; Körner, R.; McAdam, S.N.; Anthonsen, D.; Quaesten, H.; Norén, O.; Roepstorff, P.; Thorsby, E.; et al. Identification of a gliadin T-cell epitope in celiac disease: General importance of gliadin deamidation for intestinal T-cell recognition. Scand. J. Immunol. 1998, 48, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A. The characterization of the repertoire of wheat antigens and peptides involved in the humoral immune responses in patients with gluten sensitivity and Crohn’s disease. ISRN Allergy 2011, 2011, 950104. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, I.N.; Sipahi, A.M.; Damião, A.O.; de Brito, T.; Cançado, E.L.R.; Leser, P.G.; Laudanna, A.A. Celiac disease in Brazilian adults. J. Clin. Gasroenterol. 2002, 34, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Gillett, P.M.; Gillett, H.R.; Israel, D.M.; Metzger, D.L.; Stewart, L.; Chanoine, J.P.; Freeman, H.J. High prevalence of celiac disease in patients with type 1 diabetes detected by antibodies to endomysium and tissue transglutaminase. Can. J. Gastorenterol. 2001, 15, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Counsell, C.E.; Taha, A.; Ruddell, W.S. Celiac disease and autoimmune thyroid disease. Gut 1994, 35, 844–846. [Google Scholar] [CrossRef]
- Reinke, Y.; Behrendt, M.; Schmidt, S.; Zimmer, K.P.; Naim, H.Y. Impairment of protein trafficking by direct interaction of gliadin peptides with actin. Exp. Cell Res. 2011, 317, 2124–2135. [Google Scholar] [CrossRef]
- Sugai, E.; Cherñavsky, A.; Pedreira, S.; Smecuol, E.; Vazquez, H.; Niveloni, S.; Mazure, R.; Mauriro, E.; Rabinovich, G.A.; Bai, J.C. Bone-specific antibodies in sera from patients with celiac disease: Characterization and implications in osteoporosis. J. Clin. Immunol. 2002, 22, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Cuoco, L.; Chimenti, C.; Pieroni, M.; Fioravanti, G.; Gentiloni, N.; Maseri, A.; Gasbarrini, G. Celiac disease associated with autoimmune myocarditis. Circulation 2002, 105, 2611–2618. [Google Scholar] [CrossRef]
- Sárdy, M.; Kárpáti, S.; Merkl, B.; Paulsson, M.; Smyth, N. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J. Exp. Med. 2002, 195, 747–757. [Google Scholar] [CrossRef]
- Stamnaes, J.; Dorum, S.; Fleckenstein, B.; Aeschlimann, D.; Sollid, L.M. Gluten T cell epitope targeting by TG3 and TG6: Implications for dermatitis herpertiformis and gluten ataxia. Amino Acids 2010, 39, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Sanders, D.S.; Woodroofe, N.; Williamson, C.; Grünewald, R.A. Gluten ataxia. Cerebellum 2008, 7, 494–498. [Google Scholar] [CrossRef]
- Deconinck, N.; Scaillon, M.; Segers, V.; Groswasser, J.J.; Dan, B. Opsoclonusmyoclonus associated with celiac disease. Pediatr. Neurol. 2006, 34, 312–314. [Google Scholar] [CrossRef]
- Pereira, A.C.; Edwards, M.J.; Buttery, P.C.; Hawkes, C.H.; Quinn, N.P.; Giovannoni, G.; Hadjivasasiliou, M.; Bhatia, K.P. Choreic syndrome and celiac disease: A hitherto unrecognised association. Mov. Disord. 2004, 19, 478–482. [Google Scholar] [CrossRef]
- Schrödl, D.; Kahlenberg, F.; Peter-Zimmer, K.; Hermann, W.; Kühn, H.J.; Mothes, T. Intrathecal synthesis of autoantibodies against tissue transglutaminase. J. Autoimmun. 2004, 22, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Sanders, D.S.; Grünewald, R.A.; Woodroofe, N.; Boscolo, S.; Aeschlimann, D. Gluten sensitivity: From gut to brain. Lancet Neurol. 2010, 9, 318–330. [Google Scholar] [CrossRef]
- Smith, K.M.; Davidson, J.M.; Garside, P. T-cell activation occurs simultaneously in local and peripheral lymphoid tissue following oral administration of a range of doses of immunogenic or tolerogenic antigen although tolerized T cells display a defect in cell division. Immunology 2002, 106, 144–158. [Google Scholar] [CrossRef]
- Shor, D.B.; Barzilai, O.; Ram, M.; Izhaky, D.; Porat-Katz, B.S.; Chapman, J.; Blank, M.; Anaya, J.M.; Shoenfeld, Y. Gluten sensitivity in multiple sclerosis: Experimental myth or clinical truth? Ann. N. Y. Acad. Sci. 2009, 1173, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Zarei, M.; Kenton, A.; Allroggen, H. Gluten sensitivity and neuromyelitis optica: Two case reports. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1028–1030. [Google Scholar] [CrossRef] [PubMed]
- Katz, Y.; Goldberg, M.R.; Zadik-Mnuhin, G.; Leshno, M.; Heyman, E. Cross-sensitization between milk proteins: Reactivity to a “kosher” epitope? Isr. Med. Assoc. J. 2008, 10, 85–88. [Google Scholar] [PubMed]
- Kristjánsson, G.; Venge, P.; Hällgren, R. Mucosal reactivity to cow’s milk protein in celiac disease. Clin. Exp. Immunol. 2007, 147, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Triolo, G.; Accardo-Palumbo, A.; Dieli, F.; Ciccia, F.; Ferrante, A.; Giardina, E.; Licata, G. Humoral and cell mediated immune response to cow’s milk proteins in Behçet’s disease. Ann. Rheum. Dis. 2002, 61, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Triolo, G.; Triolo, G.; Accardo-Palumbo, A.; Carbone, M.C.; Giardina, E.; LaRocca, G. Behçet’s disease and celiac disease. Lancet 1995, 346, 1495. [Google Scholar] [CrossRef]
- Kolb, H.; Pozzilli, P. Cow’s milk and type 1 diabetes: The gut immune system deserves attention. Immunol. Today 1999, 20, 108–110. [Google Scholar] [CrossRef]
- Vaarala, O.; Klemetti, P.; Savilahti, E.; Reijonen, H.; Ilonen, J.; Akerblom, H.K. Cellular immune response to cow’s milk beta-lactoglobulin in patients with newly diagnosed IDDM. Diabetes 1996, 45, 178–182. [Google Scholar] [CrossRef]
- Virtanen, S.M.; Läärä, E.; Hyppönen, E.; Reijonen, H.; Räsänen, L.; Aro, A.; Knip, M.; Ilonen, J.; Akerblom, H.K. Cow’s milk consumption, HLA-DQB1 genotype, and type 1 diabetes: A nested case-control study of siblings of children with diabetes: Childhood diabetes in Finland study group. Diabetes 2000, 9, 912–917. [Google Scholar]
- Virtanen, S.M.; Nevalainen, J.; Kronberg-Kippilä, C.; Ahonen, S.; Tapanainen, H.; Uusitalo, L.; Takkinen, H.M.; Ninistö, S.; Ovaskainen, M.L.; Kenward, M.G. Food consumption and advanced beta cell autoimmunity in young children with HLA-conferred susceptibility to type 1 diabetes: A nested case-control design. Am. J. Clin. Nutr. 2012, 95, 471–478. [Google Scholar] [CrossRef]
- Stefferl, A.; Schubart, A.; Storch, M.; Amini, A.; Mather, I.; Lassman, H.; Linington, C. Butyrophilin, a milk protein, modulates the encephalitogenic T cell response to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis. J. Immunol. 2000, 165, 2859–2865. [Google Scholar] [CrossRef]
- Vojdani, A.; Campbell, A.W.; Anyanwu, E.; Kashanian, A.; Bock, K.; Vojdani, E. Antibodies to neuron-specific antigens in children with autism: Possible cross-reaction with encephalitogenic proteins from milk. Chlamydia pneumonia and Streptococcus group A. J. Neuroimmunol. 2002, 129, 168–177. [Google Scholar] [CrossRef]
- Guggenmos, J.; Schubart, A.S.; Ogg, S.; Andersson, M.; Olsson, T.; Mather, I.H.; Linington, C. Antibody cross-reactivity between myelin oligodendrocyte glycoprotein and the milk protein butyrophilin in multiple sclerosis. J. Immunol. 2004, 172, 661–668. [Google Scholar] [CrossRef]
- Riemekasten, G.; Marell, J.; Hentschel, C.; Klein, R.; Burmester, G.R.; Schoessler, W.; Hiepe, F. Casein is an essential cofactor in autoantibody reactivity directed against the C-terminal SmD1 peptide AA 83-119 in systemic lupus erythematosus. Immunobiology 2002, 206, 537–545. [Google Scholar] [CrossRef]
- Sletten, G.B.; Halvorsen, R.; Egaas, E.; Halstensen, T.S. Casein-specific immunoglobulins in cow’s milk allergic patient subgroups reveal a shift to IgA dominance in tolerant patients. Pediatr. Allergy Immunol. 2007, 18, 71–80. [Google Scholar] [CrossRef]
- Wildner, G.; Diedrichs-Mὂhring, M. Autoimmune uveitis induced by molecular mimicry of peptide from rotavirus, bovine casein and retinal S-antigen. Eur. J. Immunol. 2003, 33, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Monetini, L.; Barone, F.; Stefanini, L.; Petrone, A.; Walk, T.; Jung, G.; Thorpe, R.; Pozzilli, P.; Cavallo, M.G. Establishment of T cell lines to bovine beta-casein and beta-casein-derived epitopes in patients with type 1 diabetes. J. Endocrinol. 2003, 176, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, M.G.; Fava, D.; Monetini, L.; Barone, F.; Pozzilli, P. Cell-mediated immune response to beta casein in recent-onset insulin-dependent diabetes: Implications for disease pathogenesis. Lancet 1996, 348, 926–928. [Google Scholar] [CrossRef]
- Elian, M.; Nightingale, S.; Dean, G. Multiple sclerosis among United Kingdom-born children of immigrants from the Indian subcontinent, Africa and the West Indies. J. Neurol. Neurosurg. Psychiatry 1990, 53, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Kahana, E.; Zilber, N.; Abramson, J.H.; Biton, V.; Leibowitz, Y.; Abramsky, O. Multiple sclerosis: Genetic versus environmental aetiology: Epidemiology in Israel updated. J. Neurol. 1994, 241, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lauer, K. Diet and multiple sclerosis. Neurology 1997, 49, S55–S61. [Google Scholar] [CrossRef]
- Compston, A. Risk factors for multiple sclerosis: Race or place? J. Neurol. Neurosurg. Psychiatry 1990, 53, 821–823. [Google Scholar] [CrossRef]
- Agranoff, B.W.; Goldberg, D. Diet and the geographical distribution of multiple sclerosis. Lancet 1974, 2, 1061–1066. [Google Scholar] [CrossRef]
- Henry, J.; Miller, M.M.; Pontarotti, P. Structure and evolution of the extended B7 family. Immunol. Today 1999, 20, 285–288. [Google Scholar] [CrossRef]
- Gardinier, M.V.; Amiguet, P.; Linington, C.; Matthieu, J.M. Myelin/oligodendrocyte glycoprotein is a unique member of the immunoglobulin superfamily. J. Neurosci. Res. 1992, 33, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Jack, L.J.; Mather, I.H. Cloning and analysis of cDNA encoding bovine butyrophilin, an apical glycoprotein expressed in mammary tissue and secreted in association with the milk-fat globule membrane during lactation. J. Biol. Chem. 1990, 265, 14481–14486. [Google Scholar] [CrossRef]
- Amor, S.; Groome, N.; Linington, C.; Morris, M.M.; Dornmair, K.; Gardinier, M.V.; Matthieu, J.M.; Baker, D. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J. Immunol. 1994, 153, 4349–4356. [Google Scholar]
- Brunner, C.; Lassmann, H.; Waehneldt, T.V.; Matthieu, J.M.; Linington, C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2’, 3’-cyclic nucleotide 3’-phosphodiesterase in the CNS of adult rats. J. Neurochem. 1989, 52, 296–304. [Google Scholar] [CrossRef]
- Cobo-Calvo, A.; Ruiz, A.; Richard, C.; Blondel, S.; Cavagna, S.; Strazielle, N.; Ghersi-Egea, J.F.; Giraudon, P.; Marignier, R. Purified IgG from aquaporin-4 neuromyelitis optica spectrum disorder patients alters blood-brain barrier permeability. PLoS ONE 2020, 15, e0238301. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. AQP4 antibodies in neuromyelitis optica: Diagnostic and pathogenetic relevance. Nat. Rev. Neurol. 2010, 6, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Vaishnav, R.A.; Liu, R.; Chapman, J.; Roberts, A.M.; Ye, H.; Rebolledo-Mendez, J.D.; Tabira, T.; Fitzpatrick, A.H.; Achiron, A.; Running, M.P.; et al. Aquaporin 4 molecular mimicry and implications for neuromyelitis optica. J. Neuroimmunol. 2013, 260, 92–98. [Google Scholar] [CrossRef]
- Kinoshita, M.; Naatsuji, Y.; Kimura, T.; Moriya, M.; Takata, K.; Okuno, T.; Kumanogoh, A.; Kajiyama, K.; Yoshikawa, H.; Sakoda, S. Anti-aquaporin-4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen-specific T cells. Biochem. Biophys. Res. Commun. 2010, 394, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Mukherjee, P.S.; Berookhim, J.; Kharrazian, D. Detection of antibodies against human and plant aquaporins in patients with multiple sclerosis. Autoimmune Dis. 2015, 2015, 905208. [Google Scholar] [PubMed]
- Artur, M.A.S.; Zhao, T.; Ligterink, W.; Schranz, E.; Hilhorst, H.I.M. Dissecting the genomic diversification of late embryogenesis abundant (LEA) protein gene families in plants. Genome Biol. Evol. 2019, 11, 459–471. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, M.; Cheng, H.; Sun, N.; Simu Liu, S.; Li, S.; Wang, Y.; Zheng, Y.; Uversky, V.N. The effect of phosphorylation on the salt-tolerance-related functions of the soybean protein PM18, a member of the group-3 LEA protein family. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xing, M.; Yang, W.; Xmu, X.; Wang, X.; Lu, F.; Wang, Y.; Zhang, L. Genome-wide identification of and functional insights into the late embryogenesis abundant (LEA) gene family in bread wheat (Triticum aestivum). Sci. Rep. 2019, 9, 13375. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, D.; Kumar, S.; Rampuria, S.; Reddy, A.R.; Kirti, P.B. Ectopic expression of an atypical hydrophobic group 5 LEA protein from wild peanut Arachis diogoi confers abiotic stress tolerance in tobacco. PLoS ONE 2016, 11, e0150609. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Li, X. Identification and phylogenetic analysis of late embryogenesis abundant proteins family in tomato (Solanum lycopersicum). Planta 2015, 241, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Meng, B.; Chen, W.; Ge, X.; Liu, S.; Yu, J. A proteomic study on post diapaused embryonic development of brine shrimp (Artemia franciscana). Proteomics 2007, 7, 3580–3591. [Google Scholar] [CrossRef]
- Tiunova, A.A.; Anokhin, K.V.; Saha, A.R.; Schmidt, O.; Hanger, D.P.; Anderton, B.H.; Davies, A.M.; Ninkina, N.N.; Buchman, V.L. Chicken synucleins: Cloning and expression in the developing embryo. Mech. Dev. 2000, 99, 195–198. [Google Scholar] [CrossRef]
- Killinger, B.A.; Labrie, V. Vertebrate food products as a potential source of prion-like a-synuclein. NPJ Parkinsons Dis. 2017, 3, 33. [Google Scholar] [CrossRef][Green Version]
- Yuan, J.; Zhao, Y. Evolutionary aspects of the synuclein super-family and sub-families based on large-scale phylogenetic and group-discrimination analysis. Biochem. Biophys. Res. Commun. 2013, 441, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P.; et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef]
- Tian, C.; Liu, G.; Gao, L.; Soltys, D.; Pan, C.; Stewart, T.; Shi, M.; Xie, Z.; Liu, N.; Feng, T.; et al. Erythrocytic a-synuclein as a potential biomarker for Parkinson’s disease. Transl. Neurodegener. 2019, 8, 15. [Google Scholar] [CrossRef]
- Lerner, A. The intestinal luminal sources of α-synuclein: A gastroenterologist perspective. Nutr. Rev. 2021, nuab024. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rub, U.; Gai, P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which invulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Del Tredici, K.; Braak, H. Review: Sporadic Parkinson’s disease: Development and distribution of a-synuclein pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Lerner, A.; Vojdani, E. Cross-reactivity and sequence homology between alpha-synuclein and food products: A step further for Parkinson’s disease synucleinopathy. Cells 2021, 10, 1111. [Google Scholar] [CrossRef]
- Rutishauer, U.R.S.; Sachs, L. Cell-to-cell binding induced by different lectins. J. Cell Biol. 1975, 2, 247–257. [Google Scholar] [CrossRef]
- Brudner, M.; Karpel, M.; Lear, C.; Chen, L.; Yantosca, L.M.; Scully, C.; Sarraju, A.; Sokolovska, A.; Zariffard, M.; Reza, E.; et al. Lectin-dependent enhancement of ebola virus infection via soluble and transmembrane C-type lectin receptors. PLoS ONE 2013, 8, e60838. [Google Scholar] [CrossRef] [PubMed]
- Nistala, K.; Adams, S.; Cambrook, H.; Ursu, S.; Olivito, B.; de Jager, W.; Evans, J.G.; Cimaz, R.; Bajaj-Elliott, M.; Wedderburn, L.R. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. USA 2010, 107, 14751–14756. [Google Scholar] [CrossRef]
- Vojdani, A.; Erde, J. Regulatory T cells, a potent immunoregulatory target for CAM researchers, I: The ultimate antagonist. Evid. Based Complement. Alternat. Med. 2006, 3, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Erde, J. Regulatory T cells, a potent immunoregulatory target for CAM researchers, II: Modulating allergic and infectious disease pathology. Evid. Based Complement. Alternat. Med. 2006, 3, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Erde, J. Regulatory T cells, a potent immunoregulatory target for CAM researchers, III: Modulating tumor immunity, autoimmunity and alloreactive immunity. Evid. Based Complement. Alternat. Med. 2006, 3, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Garn, H.; Potaczek, D.P.; Pfefferle, P.I. The hygiene hypothesis and new perspectives—Current challenges meeting an old postulate. Front. Immunol. 2021, 12, 637087. [Google Scholar] [CrossRef]
- Bach, J.F. Revisiting the hygiene hypothesis in the context of autoimmunity. Front. Immunol. 2021, 11, 615192. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Fang, X.; Tokuno, S.; Chung, U.; Chen, X.; Dai, X.; Liu, X.; Xu, F.; Wang, B.; Peng, P. A web visualization tool using T-cell subsets as the predictor to evaluate COVID-19 patient’s severity. PLoS ONE 2020, 15, e0239695. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, 6508. [Google Scholar] [CrossRef] [PubMed]
- Pilli, D.; Zou, A.; Tea, F.; Dale, R.C.; Brilot, F. Expanding role of T cells in human autoimmune diseases of the central nervous system. Front. Immunol. 2017, 8, 652. [Google Scholar] [CrossRef] [PubMed]
- Dardalhon, V. Role of Th1 and Th17 cells in organ-specific autoimmunity. J. Autoimmun. 2011, 31, 252–256. [Google Scholar] [CrossRef]
- Strachan, D.P. Hay fever, hygiene, and household size. Br. Med. J. 1989, 299, 1259–1260. [Google Scholar] [CrossRef]
- Strachan, D.P. Family size, infection and atopy: The first decade of the “hygiene hypothesis”. Thorax 2000, 55 (Suppl. 1), S2–S10. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 2002, 347, 911–920. [Google Scholar] [CrossRef]
- Greenwood, B.M.; Herrick, E.M.; Voller, A. Suppression of autoimmune disease in NZB and (NZB x NZW) F1 hybrid mice by infection with malaria. Nature 1970, 226, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.Y.; Ramirfez, Z.E.; Surana, N.K. A modern-world view of host-microbiota-pathogen interactions. J. Immunol. 2021, 207, 1710–1718. [Google Scholar] [CrossRef] [PubMed]
- Nagler, C.R. Modern world influences on the microbiome and their consequences for immune-mediated disease. J. Immunol. 2021, 207, 1695–1696. [Google Scholar] [CrossRef] [PubMed]
- Kharrazian, D.; Herbert, M.; Vojdani, A. Detection of islet cell immune reactivity with low glycemic index foods: Is this a concern for type 1 diabetes? J. Diab. Res. 2017, 2017, 4124967. [Google Scholar] [CrossRef] [PubMed]
- Kharrazian, D.; Herbert, M.; Vojdani, A. Immunological reactivity using monoclonal and polyclonal antibodies of autoimmune thyroid target sites with dietary proteins. J. Thyroid Res. 2017, 2017, 44354723. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environmental Factor | Associated Disease | Reference and Number |
|---|---|---|
| Crystalline silica | RA, SLE, SSc, UC | Pollard et al. [55] |
| Smoking | RA, SLE, MS, TA, IBD | Pollard et al. [56] |
| Solvents | SSc, MS | Miller et al. [57] |
| Cosmetics | RA, SLE, PBC | Pollard et al. [58] |
| Mercury | TA | Gallagher et al. [70] |
| Somers et al. [71] | ||
| Mercury | Nephrotic syndrome | Miller et al. [72] |
| Food coloring | Rising incidence of autoimmune | Lerner and Matthias [33] |
| disease | ||
| Food coloring | ADD | Carter [95] |
| ADHD | Boris and Mandel [96] | |
| Hypersensitivity | Weliky and Heiner [103] | |
| Wheat gluten and nongluten | CD | Arentz-Hansen et al. [120] |
| proteins and peptides | NCGS, Crohn’s disease | Vojdani [126] |
| Autoimmune thyroid disease | Counsell et al. [129] | |
| Osteoporosis | Sugai et al. [131] | |
| Autoimmune myocarditis | Frustaci et al. [132] | |
| Dermatitis herpetiformis | Sárdy et al. [133] | |
| Gluten ataxia | Hadjivassiliou et al. [135] | |
| Choreic syndrome | Pereira et al. [137] | |
| MS | Shor et al. [141] | |
| NMO | Jacob et al. [142] | |
| Alzheimer’s disease | Vojdani [28] | |
| Vojdani and Vojdani [29] | ||
| Milk, caseins, alpha and beta | Type 1 diabetes | Virtanen et al. [149] |
| lactalbumin | EAE | Stefferl et al. [151] |
| Autism | Vojdani et al. [152] | |
| MS | Guggenmos et al. [153] | |
| SLE | Riemekasten et al. [154] | |
| Uveitis | Wildner and Diedrichs- | |
| Môhring | ||
| Alzheimer’s disease | Vojdani [28] | |
| Vojdani and Vojdani [29] | ||
| Aquaporins from human, | NMO | Jarius and Wildemann [170] |
| tomato, corn, soy, spinach | Vaishnav et al. [171] | |
| MS | Vojdani et al. [174] | |
| α-synuclein-containing food | PD | Hawkes et al. [187] Vojdani et al. [189] |
| Lectins and agglutinins | Autoimmune diseases | Vojdani [26] |
| Vojdani et al. [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vojdani, A.; Vojdani, E. The Role of Exposomes in the Pathophysiology of Autoimmune Diseases I: Toxic Chemicals and Food. Pathophysiology 2021, 28, 513-543. https://doi.org/10.3390/pathophysiology28040034
Vojdani A, Vojdani E. The Role of Exposomes in the Pathophysiology of Autoimmune Diseases I: Toxic Chemicals and Food. Pathophysiology. 2021; 28(4):513-543. https://doi.org/10.3390/pathophysiology28040034
Chicago/Turabian StyleVojdani, Aristo, and Elroy Vojdani. 2021. "The Role of Exposomes in the Pathophysiology of Autoimmune Diseases I: Toxic Chemicals and Food" Pathophysiology 28, no. 4: 513-543. https://doi.org/10.3390/pathophysiology28040034
APA StyleVojdani, A., & Vojdani, E. (2021). The Role of Exposomes in the Pathophysiology of Autoimmune Diseases I: Toxic Chemicals and Food. Pathophysiology, 28(4), 513-543. https://doi.org/10.3390/pathophysiology28040034