
Current Progress of EMT: A New Direction of Targeted Therapy for Colorectal Cancer with Invasion and Metastasis

Abstract
:1. Colorectal Cancer
1.1. CRC Treatment
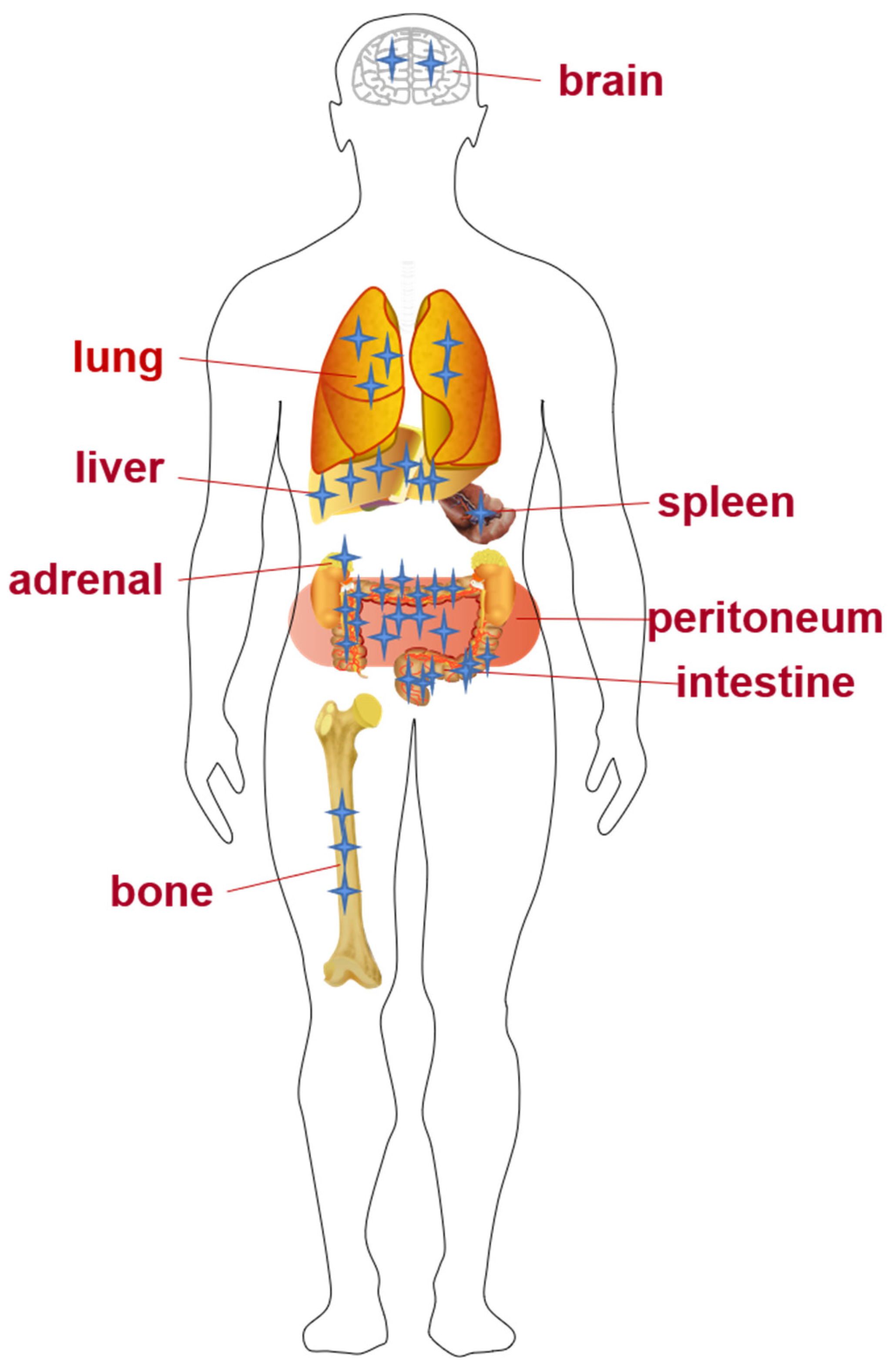
1.2. Invasion and Metastasis of CRC
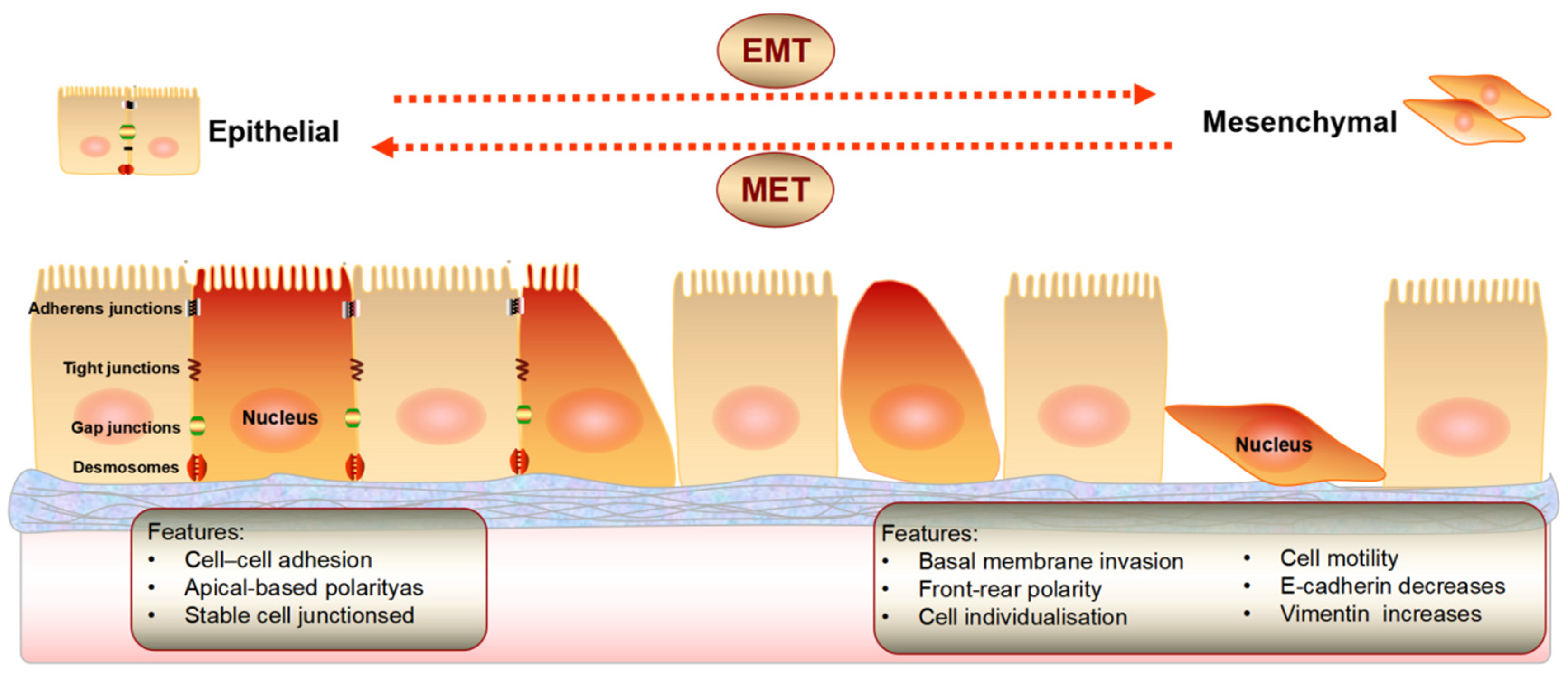
2. Epithelial Mesenchymal Transition
3. EMT in CRC Invasion and Metastasis
4. Targeted Therapy Associated with EMT
4.1. EMT and Genes
4.1.1. EMT and miRNA
4.1.2. EMT and lncRNA
4.2. EMT and Proteins
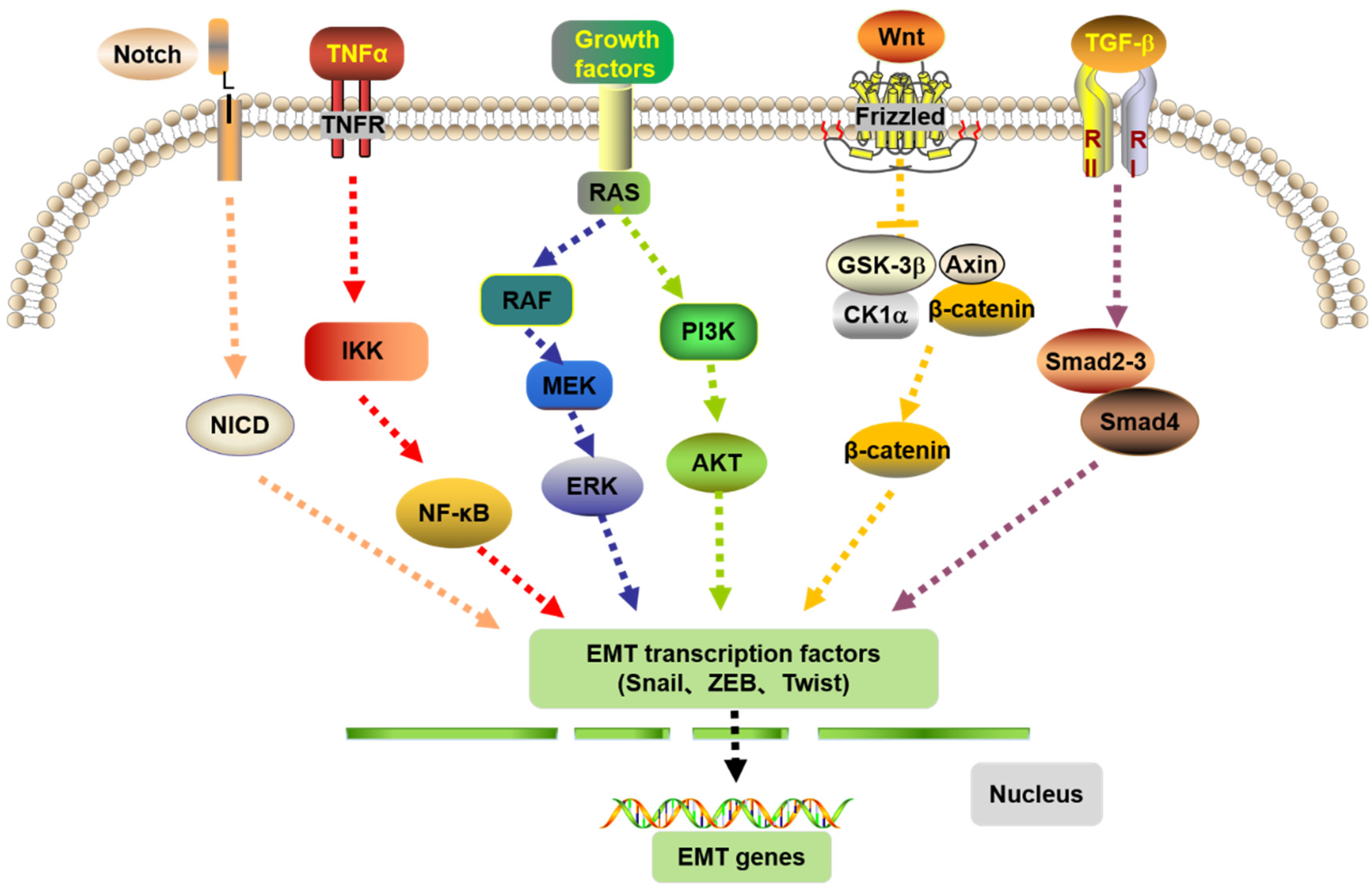
4.3. EMT and the Signaling Pathways
4.4. EMT and the Correlated Factors
4.5. EMT and MET
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| WHO | World Health Organization |
| CRC | Colorectal cancer |
| EMT | epithelial-mesenchymal transition |
| MET | mesenchymal-epithelial transition |
| TGF-β | transforming growth factor-β |
| Smad | drosophila mothers against decapentaplegic |
| Wnt | Wingless-type |
| E-cadherin | epithelial cadherin |
| STAT3 | Signal transducer and activator of transcription 3 |
| NF-κB | Notch and nuclear factor-κB |
| PI3K | phosphatidylinositol 3-kinase |
| AKT | protein kinase B |
| mTOR | mammalian target of rapamycin |
| MAPK | mitogen-activated protein kinase |
| FAK | Focal Adhesion Kinase |
| EGF | epidermal growth factor |
| FGF | Fibroblast Growth Factor |
| VEGF | vascular endothelial growth factor |
| ZEB | zinc-finger E-box-binding |
| c-Met | c-Mesenchymal-epithelial transition factor |
| ERK | extracellular signal-regulated protein kinase |
| miR | micro RNA |
| let-7a | let-7amimics |
| HDACs | histone deacetylases |
| RKIP | raf kinase inhibitory protein |
| DKK1 | Dickkopf-1 |
| PSK | polysaccharide-K |
| LHPP | Phospholysine phosphohistidine inorganic pyrophosphate phos phatase |
References
- Mattiuzzi, C.; Sanchis-Gomar, F.; Lippi, G. Concise update on colorectal cancer epidemiology. Ann. Transl. Med. 2019, 7, 609. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, R. Advances in studying lung metastases in colorectal cancer. Chongqing Med. 2021, 50, 689–693. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuge, C.; Liu, S. Progress in epithelial stromal transformation in rectal cancer. Gastroenterology 2016, 21, 307–310. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, S. Progress in the clinical treatment of colorectal cancer. New Clin. Med. China 2018, 11, 202–208. [Google Scholar] [CrossRef]
- Jiang, W.; Liu, J. Chemotherapeutic progression in colorectal cancer. China Cancer 2011, 20, 200–203. Available online: https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFD2011&filename=ZHLU201103018&uniplatform=NZKPT&v=IP3zsjdlzfD3KS0n14fi8fhu6zhbhaRJpHa0xnnSsyG__SdD-f9wisGe8eoDdb_6 (accessed on 14 October 2022).
- Du, N. Advances in colorectal cancer treatment. Chin. J. Cancer Prev. Treat. 2017, 9, 350–355. [Google Scholar] [CrossRef]
- Tan, S.; Hu, J.; Rao, S.; Ouyang, L.; Pan, Q.; Yi, P.; Cao, D.; Li, J.; Chen, X.; Yang, L.; et al. EMT-mediated resistance to tumor therapy. Chem. Life 2021, 41, 857–869. [Google Scholar] [CrossRef]
- Wang, S.Y.; Liu, S.; Liu, Y.Y.; Guo, M.L.; Zhang, S.Y.; An, Q.; Jiang, Y.X. Analysis of differential expression and prognostic-related genes in colorectal cancer. Tumor Prev. Ther. 2021, 34, 509–519. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, L.; Hong, Y. Research Progression of Tumor Microenvironment and Tumor Cell Metastasis; Digestive Diseases Society of Guizhou Medical Association: Zunyi, China, 2014. [Google Scholar] [CrossRef]
- Karthika, C.; Hari, B.; Rahman, M.; Akter, R.; Najda, A.; Albadrani, G.; Sayed, A.; Akhtar, M.; Abdel-Daim, M. Multiple strategies with the synergistic approach for addressing colorectal cancer. Biomed. Pharmacother. 2021, 140, 111704. [Google Scholar] [CrossRef]
- Li, A.; Tan, Z.; Fu, C. Analysis of risk factors for bone metastases within 5 years after radical resection of colorectal cancer. Chin. J. Gastrointest. Surg. 2017, 20, 58–61. [Google Scholar] [PubMed]
- Benson, A.B.; Venook, A.P.; Cederquist, L.; Chan, E.; Chen, Y.J.; Cooper, H.; Deming, D.; Engstrom, P.; Enzinger, P.C.; Fichera, A.; et al. Colon Cancer, Version 1.2017, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 2017, 15, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Zarour, L.; Anand, S.; Billingsley, K.; Bisson, W.H.; Cercek, A.; Clarke, M.F.; Coussens, L.M.; Gast, C.E.; Geltzeiler, C.B.; Hansen, L.; et al. Colorectal Cancer Liver Metastasis: Evolving Paradigms and Future Directions. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Guan, X.; Wang, X. Research status and treatment of bone metastases in colorectal cancer. Chin. Electron. J. Color. Dis. 2020, 9, 605–609. [Google Scholar] [CrossRef]
- Byrne, B.; Geddes, T.; Welsh, F.; John, T.G.; Chandrakumaran, K.; Rees, M. The incidence and outcome of brain metastases after liver resection for colorectal cancer metastases. Colorectal Dis. 2012, 14, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, R.; Covotta, F.; Aucello, A.; Montalto, G.; Procacciante, F.; Marcheggiani, A.; Covotta, A. Surgical treatment of isolated lung and adrenal metastasis from colorectal cancer. Ann. Ital. Chir. 2012, 83, 337–342. [Google Scholar]
- Abi Saad, G.S.; Hussein, M.; El-Saghir, N.S.; Termos, S.; Sharara, A.I.; Shamseddine, A. Isolated splenic metastasis from colorectal cancer. Int. J. Clin. Oncol. 2011, 16, 306–313. [Google Scholar] [CrossRef]
- Zhou, Y.; Mao, D. Progress in the correlation between metastasis in breast cancer and EMT/MET. World’s Lest Med. Inf. Abstr. 2018, 18, 56–58. [Google Scholar] [CrossRef]
- Myong, N.H. Loss of E-cadherin and Acquisition of Vimentin in Epithelial-Mesenchymal Transition are Noble Indicators of Uterine Cervix Cancer Progression. Korean J. Pathol. 2012, 46, 341–348. [Google Scholar] [CrossRef]
- Zhu, Q.C.; Gao, R.Y.; Wu, W.; Qin, H.L. Epithelial-mesenchymal transition and its role in the pathogenesis of colorectal cancer. Asian Pac. J. Cancer Prev. 2013, 14, 2689–2698. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Hsu, T.I.; Chang, Y.C.; Chan, M.H.; Lu, P.J.; Hsiao, M. Stationed or Relocating: The Seesawing EMT/MET Determinants from Embryonic Development to Cancer Metastasis. Biomedicines 2021, 9, 1265. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; He, Z.; Bai, S. Advances of EMT-related LncRNA in colorectal cancer. J. Pract. Cancer 2019, 34, 1911–1914. [Google Scholar] [CrossRef]
- Nieto, A.M. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 9, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwitalla, S.; Ziegler, P.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Liu, L.; Zhou, X. Progress in epithelial interstiization-related transcriptional regulation and signaling pathways in colorectal cancer. Shandong Med. 2016, 56, 96–98. [Google Scholar] [CrossRef]
- Ahmadiankia, N.; Khosravi, A. Significance of epithelial-to-mesenchymal transition inducing transcription factors in predicting distance metastasis and survival in patients with colorectal cancer: A systematic review and meta-analysis. J. Res. Med. Sci. 2020, 25, 60. [Google Scholar] [CrossRef]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Aigner, K.; Dampier, B.; Descovich, L.; Mikula, M.; Sultan, A.; Schreiber, M.; Mikulits, W.; Brabletz, T.; Strand, D.; Obrist, P.; et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007, 26, 6979–6988. [Google Scholar] [CrossRef]
- Zhao, P.; Lu, Y.; Yang, J. Progress in the pathways related to epithelial stromal transformation and its targeted therapy in colorectal cancer. Int. J. Dig. Dis. 2020, 40, 104–108. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang RY, J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.; Bai, X. The role of EMT in cancer metastasis: An endoenvironment-dependent regulatory process. Chem. Life 2021, 41, 428–436. [Google Scholar] [CrossRef]
- Hou, B.; Li, W.; Xia, P.; Zhao, F.; Liu, Z.; Zeng, Q.; Wang, S.; Chang, D. LHPP suppresses colorectal cancer cell migration and invasion in vitro and in vivo by inhibiting Smad3 phosphorylation in the TGF-β pathway. Cell Death Discov. 2021, 7, 273. [Google Scholar] [CrossRef] [PubMed]
- Elsum, I.A.; Martin, C.; Humbert, P.O. Scribble regulates an EMT polarity pathway through modulation of MAPK-ERK signaling to mediate junction formation. J. Cell Sci. 2013, 126, 3990–3999. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Song, S.H.; Choi, Y.H.; Hwang, K.H.; Yun, J.S.; Song, H.; Cha, S.Y.; Cho, S.B.; Lee, I.; Kim, H.S.; et al. Competing Endogenous RNA of Snail and Zeb1 UTR in Therapeutic Resistance of Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 9589. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Yu, J.; Yu, H. Chemoresistance to doxorubicin induces epithelial-mesenchymal transition via upregulation of transforming growth factor β signaling in HCT116 colon cancer cells. Mol. Med. Rep. 2015, 12, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, H.; Miyoshi, N.; Nagai, K.; Tomimaru, Y.; Nagano, H.; Sekimoto, M.; Doki, Y.; Mori, M.; Ishii, H. Epithelial-mesenchymal transition with expression of SNAI1-induced chemoresistance in colorectal cancer. Biochem. Biophys. Res. Commun. 2009, 390, 1061–1065. [Google Scholar] [CrossRef]
- Shen, L.; Qu, X.; Ma, Y.; Zheng, J.; Chu, D.; Liu, B.; Li, X.; Wang, M.; Xu, C.; Liu, N.; et al. Tumor suppressor NDRG2 tips the balance of oncogenic TGF-β via EMT inhibition in colorectal cancer. Oncogenesis 2014, 3, e86. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaderna, S.; Schmalhofer, O.; Hlubek, F.; Berx, G.; Eger, A.; Merkel, S.; Jung, A.; Kirchner, T.; Brabletz, T. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology 2006, 131, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kang, Y. Probing the Fifty Shades of EMT in Metastasis. Trends Cancer 2016, 2, 65–67. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells-an integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Bi, X.F.; Guo, L.; Li, W.B.; Xu, Z.J. Molecular pathology epidemiology of KRAS and BRAF gene mutations in colorectal cancer. Chin. J. Cancer Prev. Treat. 2021, 28, 805–810. [Google Scholar] [CrossRef]
- Yang, Y.; Feng, M.; Bai, L.; Liao, W.; Zhou, K.; Zhang, M.; Wu, Q.; Wen, F.; Lei, W.; Zhang, P.; et al. Comprehensive analysis of EMT-related genes and lncRNAs in the prognosis, immunity, and drug treatment of colorectal cancer. J. Transl. Med. 2021, 19, 391. [Google Scholar] [CrossRef]
- Liu, Z.; Wei, E.; Lu, Y. Progress in studying the role of epithelial-stromal transformation in tumor development. J. Zhejiang Univ. (Med. Ed.) 2015, 44, 211–216. Available online: https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFDTEMP&filename=ZJYB201502021&uniplatform=NZKPT&v=vIHNok4MEzNpBAh5cfW1skJQssnh-Y7TKJX9IPItWsPKCtAcvXyxHbK7MLL-6O6y (accessed on 14 October 2022).
- Zuo, K.; Li, Y. Progress in colorectal cancer-related microRNAs. Chin. J. Oncol. Surg. 2018, 10, 57–59. [Google Scholar] [CrossRef]
- Ouyang, W.; Wu, X.D.; Chen, Q.; Chen, X.L.; Zhang, P.H.; Yang, H.M. Progress in studying long noncoding RNA and epithelial stromal transformation in colorectal cancer. Shandong Med. 2018, 58, 116–119. [Google Scholar] [CrossRef]
- Peng, K.; Chen, H. Role of epithelial stromal cell transformation in tumor invasion, metastasis and chemoresistance. J. Pract. Med. 2018, 35, 85–87. [Google Scholar] [CrossRef]
- Shen, X.; Jiang, H.; Chen, Z.; Lu, B.; Zhu, Y.; Mao, J.; Chai, K.; Chen, W. MicroRNA-145 Inhibits Cell Migration and Invasion in Colorectal Cancer by Targeting TWIST. OncoTargets Ther. 2019, 12, 10799–10809. [Google Scholar] [CrossRef] [PubMed]
- Rokavec, M.; Öner, M.G.; Li, H.; Jackstadt, R.; Jiang, L.; Lodygin, D.; Kaller, M.; Horst, D.; Ziegler, P.K.; Schwitalla, S.; et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J. Clin. Invest. 2014, 124, 1853–1867. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; He, D.; Yang, D. MiR-489 regulates chemoresistance in breast cancer via epithelial mesenchymal transition pathway. FEBS Lett. 2014, 588, 2009–2015. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Zhao, Y.; Chen, J.; Hu, J.; Wang, S.; Zhang, D.; Sun, Y. BRAF-activated long non-coding RNA contributes to colorectal cancer migration by inducing epithelial-mesenchymal transition. Oncol. Lett. 2014, 8, 869–875. [Google Scholar] [CrossRef] [Green Version]
- Amelio, I.; Bernassola, F.; Candi, E. Emerging roles of long non-coding RNAs in breast cancer biology and management. Semin. Cancer Biol. 2021, 72, 36–45. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, L.; Wang, H.; Tang, F.R.; Cai, W.Q.; Sethi, G.; Xin, H.W.; Ma, Z. Insights into Biological Role of LncRNAs in Epithelial-Mesenchymal Transition. Cells 2019, 8, 1178. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, W.; Yuan, Y.; Li, J.; Wu, J.; Yu, J.; He, Y.; Wei, Z.; Zhang, C. Long non-coding RNA H19 promotes colorectal cancer metastasis via binding to hnRNPA2B1. J. Exp. Clin. Cancer Res. 2020, 39, 141. [Google Scholar] [CrossRef]
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 2013, 26, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Moon, J.W.; Min, D.H.; Ko, E.S.; Ahn, B.; Kim, E.S.; Lee, J.Y. AHA1 regulates cell migration and invasion via the EMT pathway in colorectal adenocarcinomas. Sci. Rep. 2021, 11, 19946. [Google Scholar] [CrossRef]
- Singh, A.B.; Sharma, A.; Smith, J.J. Claudin-1 up-regulates the repressor ZEB-1 to inhibit E-cadherin expression in colon cancer cells. Gastroenterology 2011, 141, 2140–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadish, N.; Parashar, D.; Gupta, N.; Agarwal, S.; Purohit, S.; Kumar, V.; Sharma, A.; Fatima, R.; Topno, A.P.; Shaha, C.; et al. A-kinase anchor protein 4 (AKAP4) a promising therapeutic target of colorectal cancer. J. Exp. Clin. Cancer Res. 2015, 34, 142. [Google Scholar] [CrossRef]
- Liu, H.; Fu, Q.; Lu, Y.; Zhang, W.; Yu, P.; Liu, Z.; Sun, X. Anti-tubulin agent vinorelbine inhibits metastasis of cancer cells by regulating epithelial-mesenchymal transition. Eur. J. Med. Chem. 2020, 200, 112332. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Sun, B.; Liu, Z.; Li, H.; Li, H.; Gao, J.; Leng, X. Dickkopf-1 inhibits epithelial-mesenchymal transition of colon cancer cells and contributes to colon cancer suppression. Cancer Sci. 2012, 103, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Canel, M.; Serrels, A.; Miller, D.; Timpson, P.; Serrels, B.; Frame, M.C.; Brunton, V.G. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: Effects on E-cadherin dynamics. Cancer Res. 2010, 70, 9413–9422. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Hayashida, T.; Konagai, A. Direct inhibition of the transforming growth factor-β pathway by protein-bound polysaccharide through inactivation of Smad2 signaling. Cancer Sci. 2012, 103, 317–324. [Google Scholar] [CrossRef]
- Sakamoto, J.; Morita, S.; Oba, K.; Matsui, T.; Kobayashi, M.; Nakazato, H.; Ohashi, Y. Efficacy of adjuvant immunochemotherapy with polysaccharide K for patients with curatively resected colorectal cancer: A meta-analysis of centrally randomized controlled clinical trials. Cancer Immunol. Immunother. 2006, 55, 404–411. [Google Scholar] [CrossRef]
- Hu, X.; Xing, W.; Zhao, R.; Tan, Y.; Wu, X.F.; Ao, L.Q.; Li, Z.; Yao, M.W.; Yuan, M.; Guo, W.; et al. HDAC2 inhibits EMT-mediated cancer metastasis by downregulating the long noncoding RNA H19 in colorectal cancer. J. Exp. Clin. Cancer Res. 2020, 39, 270. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef]
- Brabletz, T. EMT and MET in metastasis: Where are the cancer stem cells? Cancer Cell 2012, 22, 699–701. [Google Scholar] [CrossRef] [Green Version]
- Personeni, N.; Smiroldo, V.; Giunta, E.F.; Prete, M.G.; Rimassa, L.; Bregni, G.; Sclafani, F. Tackling Refractory Metastatic Colorectal Cancer: Future Perspectives. Cancers 2021, 13, 4506. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Bader, J.S.; Braun, T.P.; Califano, A.; Clemons, P.A.; Druker, B.J.; Ewald, A.J.; Fu, H.; Jagu, S.; Kemp, C.J.; et al. An expanded universe of cancer targets. Cell 2021, 184, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Tonini, G.; Imperatori, M.; Vincenzi, B.; Frezza, A.M.; Santini, D. Rechallenge therapy and treatment holiday: Different strategies in management of metastatic colorectal cancer. J. Exp. Clin. Cancer Res. 2013, 32, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, Y.; Ying, G. Progress in epithelial stromal transformation in tumor invasion and metastasis. Zhejiang Chem. Ind. 2019, 50, 11–15. [Google Scholar] [CrossRef]
- Cao, W.; Yang, Z. Progress in tumor microenvironment and epithelial-stromal transformation in tumor metastasis. Basic Clin. Oncol. 2019, 32, 547–549. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, H.; Qiu, J. The role of epithelial-stromal transformation in tumor metastasis. China J. Biochem. Mol. Biol. 2014, 30, 1169–1175. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Zhou, H. Progress in the study of EMT and immune escape of tumor cells. Chem. Life 2017, 37, 980–985. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| References | ||||
|---|---|---|---|---|
| EMT | Regulatory proteins | E-cadherin | [9] | |
| Vimentin | ||||
| N-cadherin | ||||
| Transcription factor | Snail1 | [31] | ||
| Slug | ||||
| ZEB1 | ||||
| ZEB2 | ||||
| Twist1 | ||||
| Twist2 | ||||
| E12 | ||||
| Drug development | Molecular targeted drugs chemotherapy drugs | 5-fluorouracil | [43] | |
| Regorafenib | [73] | |||
| Bevacizumab | [14] | |||
| Cetuximab | ||||
| Panitumumab | ||||
| Molecular biomarkers | Encorafenib | [49] | ||
| Adagrasib | ||||
| miRNA | miRNA-375 | [54] | ||
| miR-145 | [55] | |||
| miR-34a | [56] | |||
| miR-200 | ||||
| miR-489 | [57] | |||
| lncRNA | BANCR | [58] | ||
| SNHG15 | ||||
| LINC01133 | ||||
| H19 | [60] | |||
| Protein | Hsp90 | [62] | ||
| AHA1 | ||||
| Claudin-1 | [63] | |||
| AKAP4 | [64] | |||
| Navelbine | [65] | |||
| Inhibitors of Wnt signaling pathway | DKK1 | [66] | ||
| Inhibitors of Src/FAK signaling pathway | PF-562, 271 | [66] | ||
| Inhibitors of TGF-β signaling pathway | PSK | [68] | ||
| LHPP | [39] | |||
| Targeted RKIP | HLX55, SHR-A1403 | [71] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Z.; Sun, W.; Li, Y.; Jiao, X.; Zhu, M.; Zhang, J.; Qing, C.; Jia, Y. Current Progress of EMT: A New Direction of Targeted Therapy for Colorectal Cancer with Invasion and Metastasis. Biomolecules 2022, 12, 1723. https://doi.org/10.3390/biom12121723
Tan Z, Sun W, Li Y, Jiao X, Zhu M, Zhang J, Qing C, Jia Y. Current Progress of EMT: A New Direction of Targeted Therapy for Colorectal Cancer with Invasion and Metastasis. Biomolecules. 2022; 12(12):1723. https://doi.org/10.3390/biom12121723
Chicago/Turabian StyleTan, Zhuomin, Wenyan Sun, Ya Li, Xingmeng Jiao, Mingliang Zhu, Junfei Zhang, Chen Qing, and Yinnong Jia. 2022. "Current Progress of EMT: A New Direction of Targeted Therapy for Colorectal Cancer with Invasion and Metastasis" Biomolecules 12, no. 12: 1723. https://doi.org/10.3390/biom12121723
APA StyleTan, Z., Sun, W., Li, Y., Jiao, X., Zhu, M., Zhang, J., Qing, C., & Jia, Y. (2022). Current Progress of EMT: A New Direction of Targeted Therapy for Colorectal Cancer with Invasion and Metastasis. Biomolecules, 12(12), 1723. https://doi.org/10.3390/biom12121723