
A Combined Chemical-Electrochemical Process to Capture CO2 and Produce Hydrogen and Electricity


Abstract
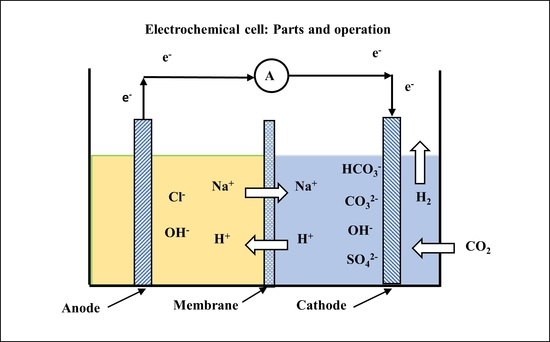
:
1. Introduction
2. Materials and Methods
2.1. General Considerations
- The evolution of hydrogen on metallic electrodes is a complex process that involves several steps. Some of them are electron transfer steps and others are catalytic in nature (Butler-Volmer, Heyrovsky, Taffel, and combinations ([20,21,22], among others)). There are several kinetic expressions depending upon which step controls the rate of the overall process. Despite the impressive amount of work carried out on this research subject many issues are still not clear. Furthermore, the kinetic behavior depends upon the electrode materials used and operating conditions ([20,21,22,23,24,25,26,27], among others). Some authors even question the existence of a redox mechanism and propose an overpotential-capacitance mechanism for the H2 electrode reaction on Pt [22]. In this work, for simplicity, we used a generic kinetic expression for the evolution of hydrogen under a galvanic cell operating conditions [22], see Section 2.6 below.
- Even for accepted mechanisms few experimental data are available for the several kinetic parameters necessary for successful simulations. In this project we used literature parameters if available and our own estimates if we could not find values in literature.
- The theory for electrical double layers (EDLs) in the case of strong electrolytes comprising monovalent ions is well developed for the case of the Gouy-Chapman-Stern model (GCS), ([28,29,30,31], among others). However, the studies published for multi-ion systems show that the theoretical treatment is very complex. In this work we introduced a Debye-Hückel linearization approximation [32] to deal with the several ionic species present, see Section 2.5 below.
- It is also a complex matter to calculate conductivities of multi-ion solutions [33], especially for high concentrations. We used approximations for these calculations, see Section 2.6 below.
2.2. Model Organization
- (1)
- Electrochemical reaction in the anode (oxidation).
- (2)
- Potential drop through the Stern layer of an electrical double layer (EDL) on the anode side.
- (3)
- Mass transfer through the diffuse layer of an electrical double layer on the anode side.
- (4)
- Mass transfer through an aqueous solution in contact with the anode.
- (5)
- Stagnant double layer in contact with the anionic exchange membrane on the anode side.
- (6)
- Restricted mass transfer through an ionic exchange membrane (IEM).
- (7)
- Stagnant double layer in contact with the anionic exchange membrane (IEM) on the cathode side.
- (8)
- Mass transfer through an aqueous solution in contact with the cathode. Inside the cathode half-cell there is also a chemical reaction between gaseous CO2 and the acidic solution. In this work we bubble in the cathode a gas mixture containing 13% CO2 V/V ratio.
- (9)
- Mass transfer through the diffuse layer of an electrical double layer on the cathode side.
- (10)
- Potential drop through the Stern layer of an EDL on the cathode side.
- (11)
- Electrochemical reaction in the cathode half-cell (reduction).
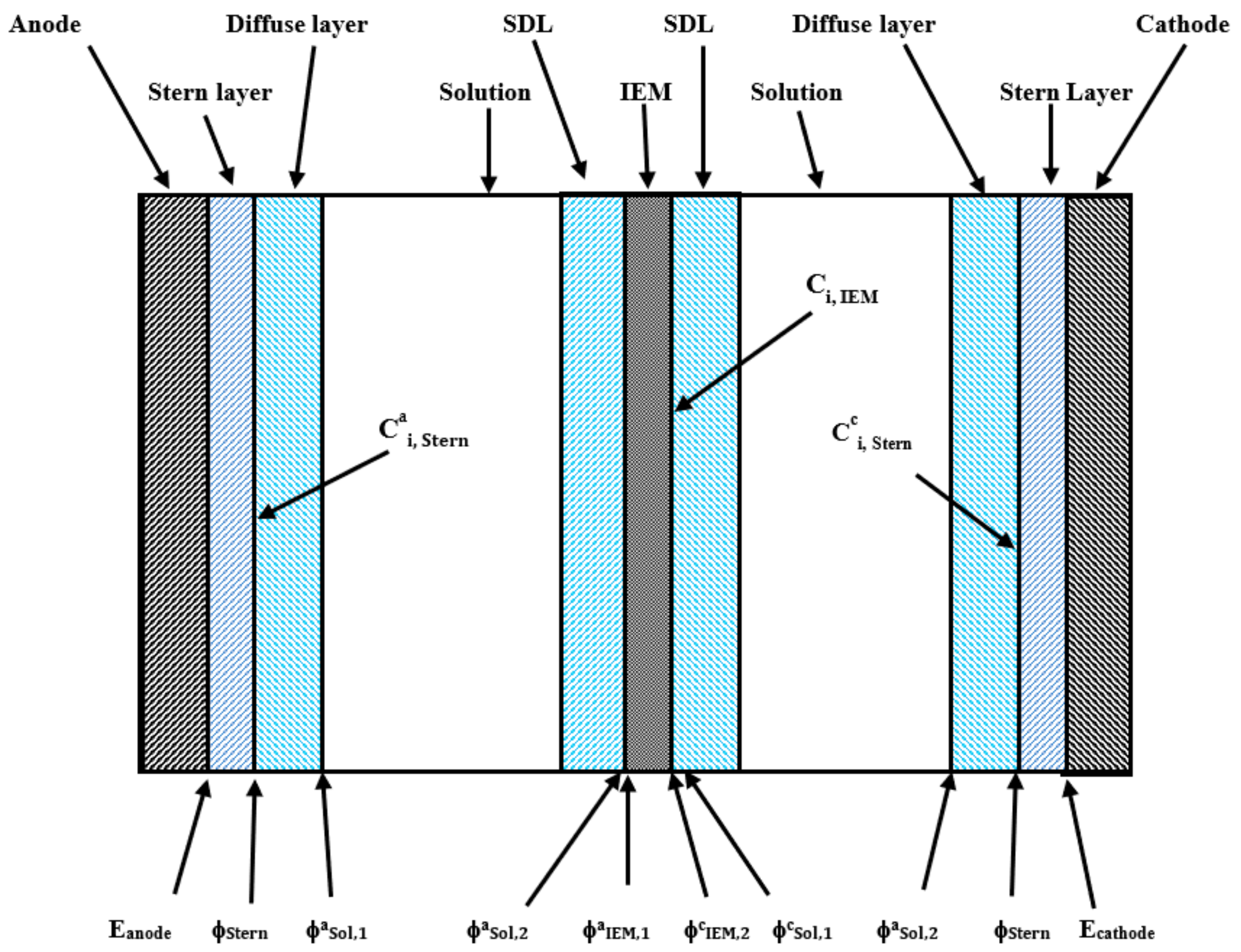
2.3. Electrode Reactions
2.4. Ion Exchange Membrane and Stagnant Diffuse Layers
2.5. Electrical Double Layers (EDLs)
2.6. Anode and Cathode Bulk Solutions
2.6.1. Anode
2.6.2. Cathode
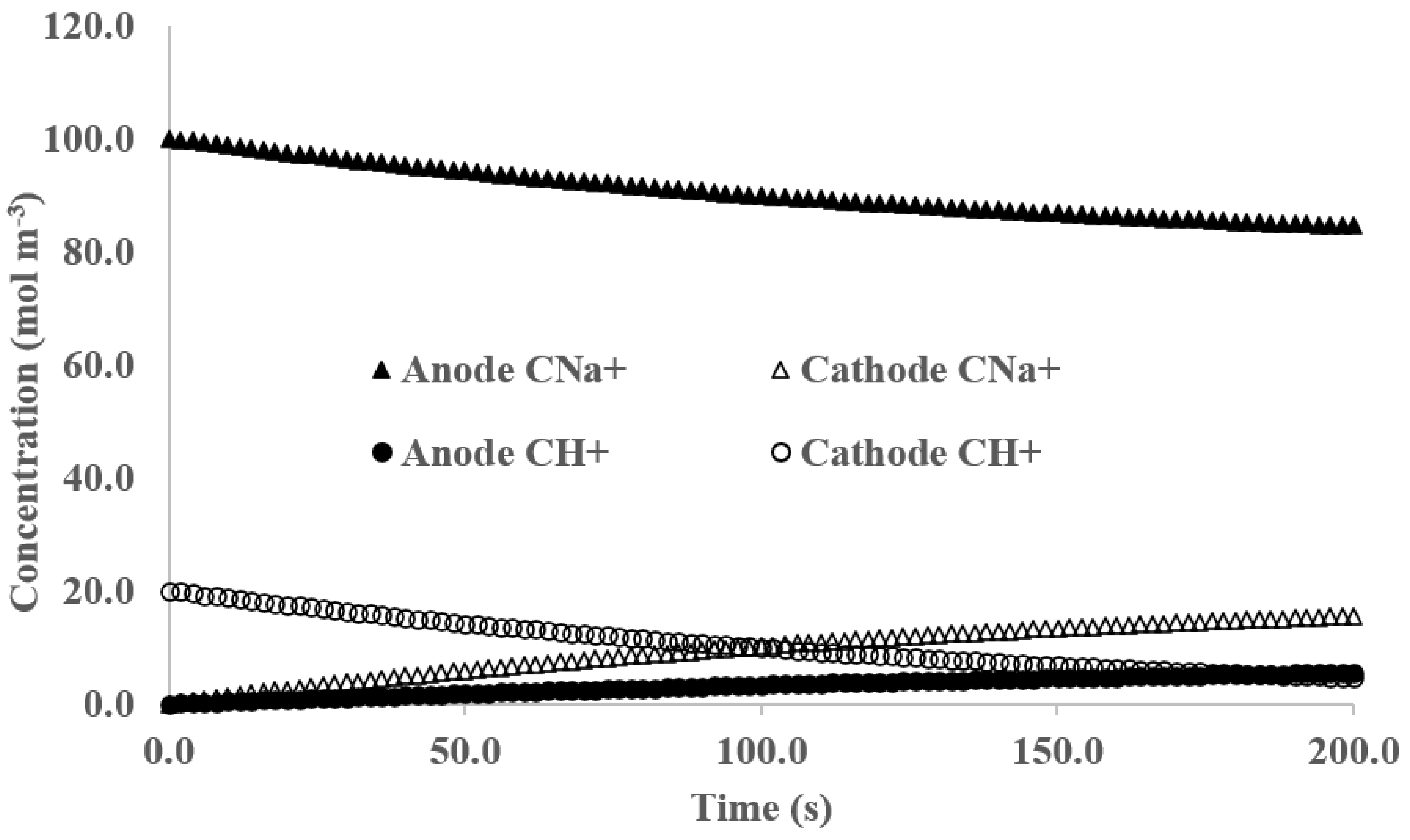
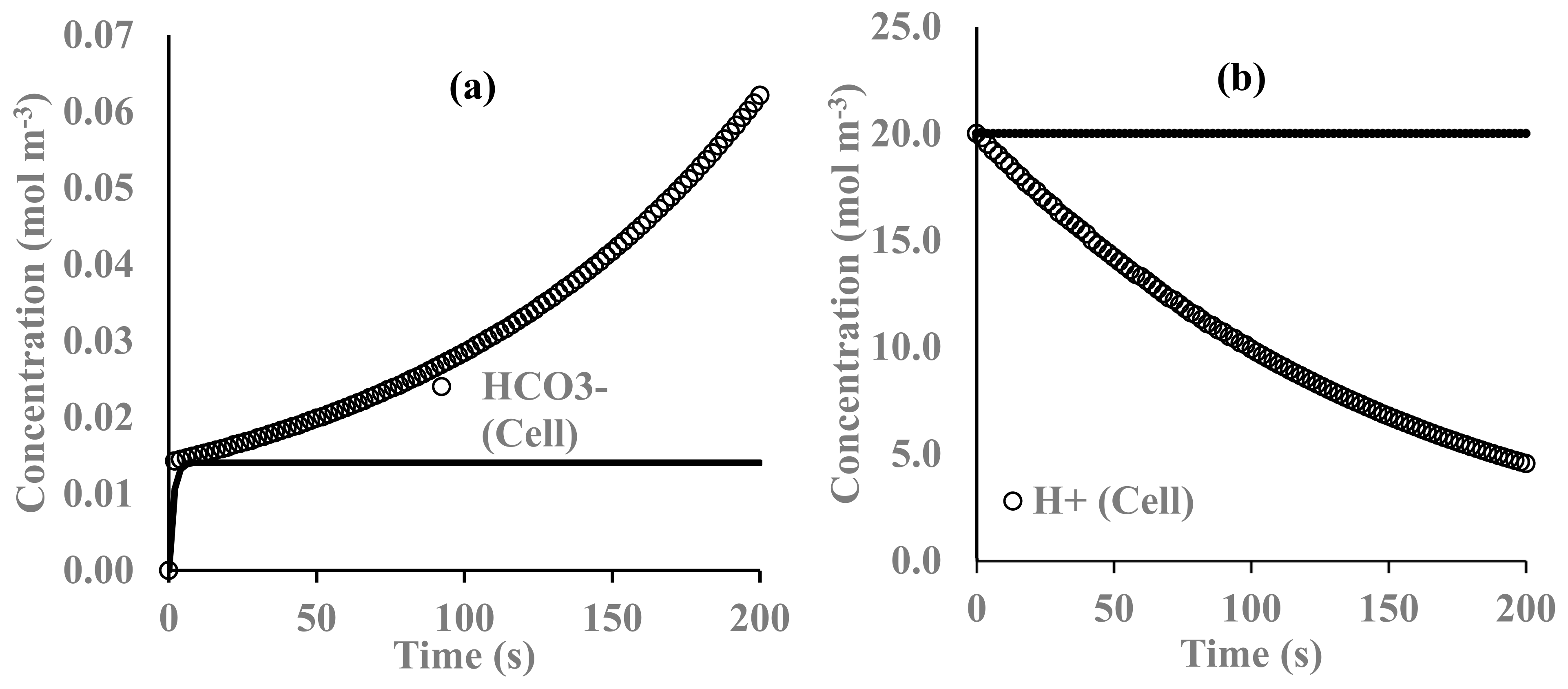
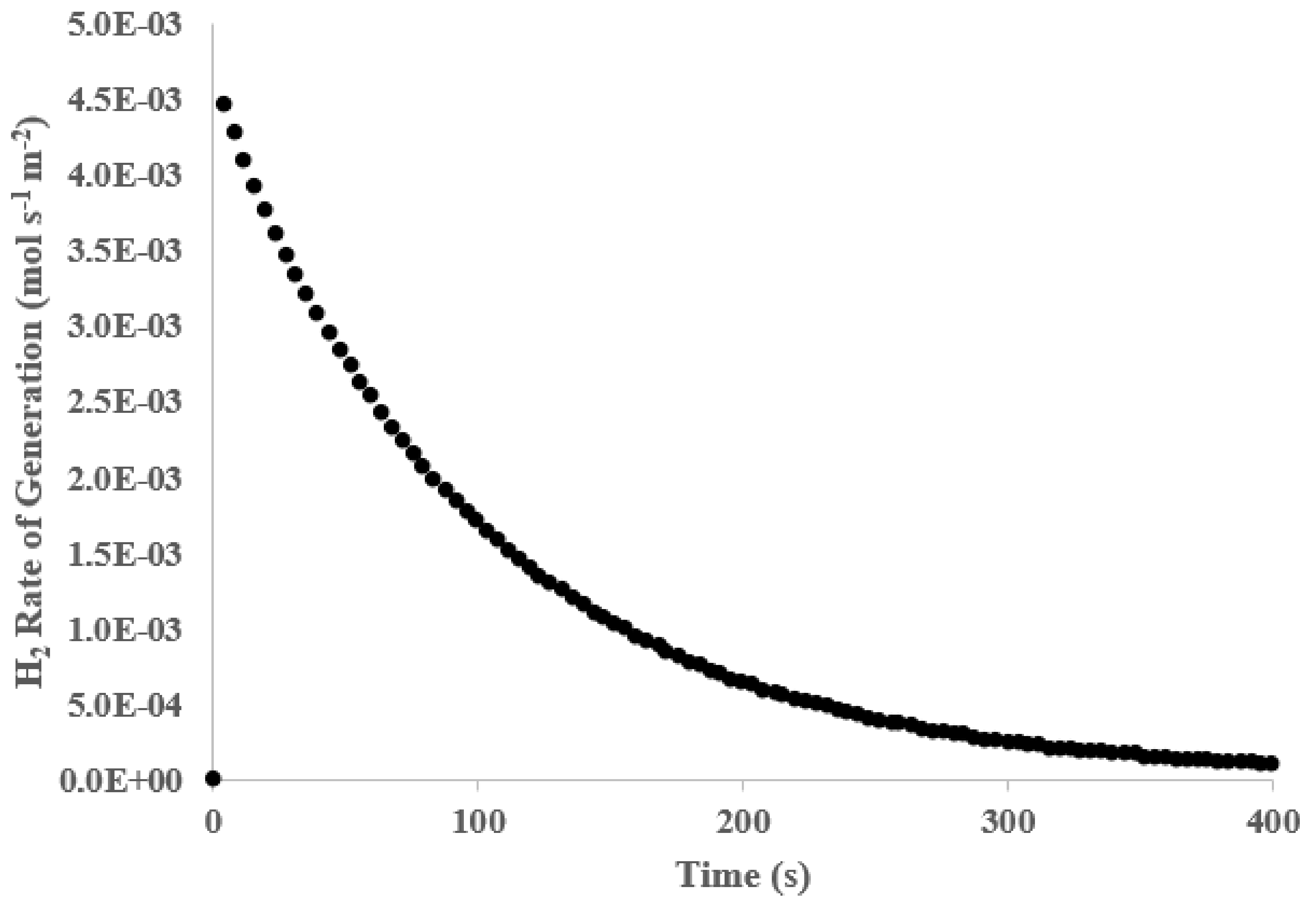
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Notation | Parameter | Value/Range | Units |
|---|---|---|---|
| Anode half-cell standard potential | −2.71 | V | |
| Cathode half-cell standard potential | 0.0 | V | |
| Anodic reaction forward constant | 6.65 × 10−4 | s−1 | |
| Cathodic reaction forward constant | 4.43 × 10−4 | s−1 | |
| Anodic reaction reverse constant | 6.65 × 10−4 | s−1 | |
| Cathodic reaction reverse constant | 6.65 × 10−4 | s−1 | |
| Initial CO2 concentration in gas phase | 0.0054 | M | |
| Initial CO2 gas phase volume fraction | 13 | % V/V | |
| Henry Constant CO2 | 0.8285 | ||
| Initial H+ cathodic concentration | 0.01–0.1 | M | |
| Initial Na+ anodic concentration | 0.1–0.01 | M | |
| Reaction 1 forward constant | 0.024 | s−1 | |
| Reaction 1 equilibrium constant | 7.31 × 10−7 | M | |
| Reaction 2 forward constant | 5.86 × 103 | s−1 M−1 | |
| Reaction 2 equilibrium constant | 4.51 × 107 | M−1 | |
| Ionic water product | 1 × 10−14 | M2 | |
| Carbonic acid second dissociation constant | 5.0 × 10−11 | M | |
| Anode length | 0.1 | m | |
| Cathode length | 0.1 | m | |
| Cell height | 0.1 | m | |
| Cell width | 0.1 | m | |
| Membrane thickness | 0.002 | m | |
| Membrane void fraction | 0.4 | (-) | |
| X | Membrane charge | 1 | M |
| w | Membrane charge sign | −1 | (-) |
| T | Temperature | 293 | K |
| Standard total pressure | 1 × 105 | Pa |
| Notation | Parameter | Value/Range | Units |
|---|---|---|---|
| H+ ion conductivity at infinite dilution | 349.8 | S cm2 mol−1 | |
| Na+ ion conductivity at infinite dilution | 50.1 | S cm2 mol−1 | |
| OH− ion conductivity at infinite dilution | 198.3 | S cm2 mol−1 | |
| Cl− ion conductivity at infinite dilution | 76.3 | S cm2 mol−1 | |
| SO42− ion conductivity at infinite dilution | 160.0 | S cm2 mol−1 | |
| HCO3− ion conductivity at infinite dilution | 44.5 | S cm2 mol−1 | |
| CO32− ion conductivity at infinite dilution | 138.6 | S cm2 mol−1 | |
| H+ ion Kohlrausch Constant | 106.92 | S cm2 L1/2 mol−3/2 | |
| Na+ ion Kohlrausch Constant | 40.55 | S cm2 L1/2 mol−3/2 | |
| OH− ion Kohlrausch Constant | 73.37 | S cm2 L1/2 mol−3/2 | |
| Cl− ion Kohlrausch Constant | 46.35 | S cm2 L1/2 mol−3/2 | |
| SO42− ion Kohlrausch Constant | 122.86 | S cm2 L1/2 mol−3/2 | |
| HCO3− ion Kohlrausch Constant | 39.31 | S cm2 L1/2 mol−3/2 | |
| CO32− ion Kohlrausch Constant | 109.92 | S cm2 L1/2 mol−3/2 |
References
- Intergovermental Panel on Climate Change (IPCC). Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2014; Available online: https://www.ipcc.ch/report/ar5/syr/ (accessed on 4 July 2021).
- Kim, Y.-H.; Park, L.K.; Yiacoumi, S.; Tsouris, C. Modular Chemical Process Intensification: A Review. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 359–380. [Google Scholar] [CrossRef]
- Timberlake, K.; Timberlake, W. Basic Chemistry, 4th ed.; Pearson: Upper Saddle River, NJ, USA, 2014; pp. 454–456. [Google Scholar]
- Greer, T. Modeling and Simulation of Post Combustion CO2 Capturing. Master’s Thesis, Faculty of Technology, Telemark University College, Porsgrunn, Norway, 2008. [Google Scholar]
- Hikita, H.; Asai, S.; Takatsuka, T. Absorption of Carbon Dioxide into Aqueous Sodium Hydroxide and Sodium Carbonate-Bicarbonate Solutions. Chem. Eng. J. 1972, 11, 131–141. [Google Scholar] [CrossRef]
- Hikita, H.; Asai, S.; Takatsuka, T. Gas absorption with a two-step instantaneous chemical reaction. Chem. Eng. J. 1972, 4, 31–40. [Google Scholar] [CrossRef]
- Li, P.-Z.; Wang, X.-J.; Liu, J.; Lim, J.S.; Zou, R.; Zhao, Y. A Triazole-Containing Metal–Organic Framework as a Highly Effective and Substrate Size-Dependent Catalyst for CO2 Conversion. J. Am. Chem. Soc. 2016, 138, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, B.; Tyo, E.C.; Seifert, S.; DeBartolo, J.; von Issendorff, B.; Zapol, P.; Vajda, S.; Curtiss, L.A. Carbon Dioxide Conversion to Methanol over Size-Selected Cu4 Clusters at Low Pressures. J. Am. Chem. Soc. 2015, 137, 8676–8679. [Google Scholar] [CrossRef] [PubMed]
- Angamuthu, R.; Byers, P.; Lutz, M.; Spek, A.L.; Bouwman, E. Electrocatalytic CO2 Conversion to Oxalate by a Copper Complex. Science 2010, 327, 313–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, D.U.; Hu, X.-M.; Daasbjerg, K.; Skrydstrup, T. Chemically and electrochemically catalysed conversion of CO2 to CO with follow-up utilization to value-added chemicals. Nat. Catal. 2018, 1, 244–254. [Google Scholar] [CrossRef]
- Xu, L.; Xiu, Y.; Liu, F.; Liang, Y.; Wang, S. Research Progress in Conversion of CO2 to Valuable Fuels. Molecules 2020, 25, 3653. [Google Scholar] [CrossRef]
- More, G.S.; Srivastava, R. Synthesis of amino alcohols, cyclic urea, urethanes, and cyclic carbonates and tandem one-pot conversion of an epoxide to urethanes using a Zn–Zr bimetallic oxide catalyst. Sustain. Energy Fuels 2021, 5, 1498–1510. [Google Scholar] [CrossRef]
- Jelmy, E.J.; Thomas, N.; Mathew, D.T.; Louis, J.; Padmanabhan, N.T.; Kumaravel, V.; John, H.; Pillai, S.C. Impact of structure, doping and defect-engineering in 2D materials on CO2 capture and conversion. React. Chem. Eng. 2021. [Google Scholar] [CrossRef]
- Bourzac, K. We Have the Technology. Nature 2017, 550, S66–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markewitz, P.; Kuckshinrichs, W.; Leitner, W.; Linssen, J.; Zapp, P.; Bongartz, R.; Schreiber, A.; Muller, T.E. World-wide Innovations in the development of Carbon Capture Technologies and the Utilization of CO2. Energy Environ. Sci. 2012, 5, 7281–7305. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Kim, J.; Joo, S.; Bu, Y.; Liu, M.; Cho, J.; Kim, G. Efficient CO2 Utilization via a Hybrid Na-CO2 System Based on CO2 Dissolution. iScience 2018, 9, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qie, L.; Lin, Y.; Connell, J.W.; Xu, J.; Dai, L. Highly Rechargeable Lithium-CO2 Batteries with a Boron- and Nitro-gen-Codoped Holey-Graphene Cathode. Angew. Chem. Int. Ed. 2017, 56, 6970–6974. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Sun, J.; Li, Z.; Zhao, Q.; Chen, C.; Chen, J. Rechargeable Room-Temperature Na-CO2 Batteries. Angew. Chem. Int. Ed. 2016, 55, 6482–6486. [Google Scholar] [CrossRef]
- Al Sadat, W.I.; Archer, L.A. The O2-assisted Al/CO2 electrochemical cell: A system for CO2 capture/conversion and electric power generation. Sci. Adv. 2016, 2, e1600968. [Google Scholar] [CrossRef] [Green Version]
- Bockris, J.O.; Mauser, H. The kinetics of the evolution and dissolution of hydrogen at electrodes. Can. J. Chem. 1959, 37, 475–488. [Google Scholar] [CrossRef]
- Radhakrishnamurthy, P.; Sathyanarayana, S.; Reddy, A.K.N. Kinetics of hydrogen evolution reaction on a stainless steel electrode. J. Appl. Electrochem. 1977, 7, 51–55. [Google Scholar] [CrossRef]
- Cheng, K.L.; Ashraf, N.; Wei, G. A New Overpotential—Capacitance Mechanism for H2 Electrode. Sensors 2006, 6, 1187–1198. [Google Scholar] [CrossRef] [Green Version]
- Rheinländer, P.J.; Herranz, J.; Durst, J.; Gasteiger, H.A. Kinetics of Hydrogen Oxidation/Evolution Reaction on Poly-crystaline Platinum in Alkaline Electrolyte Reaction Order with Respect to Hydrogen Pressure. J. Electrochem. Soc. 2014, 161, F1448–F1457. [Google Scholar] [CrossRef]
- Shinagawa, T.; Garcia-Esparza, A.T.; Takanabe, K. Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion. Sci. Rep. 2015, 5, 13801. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.T.; Liu, X.; Ji, Y.; Norskov, J.K.; Chan, K. Modeling Hydrogen Evolution Reaction Kinetics through Explicit Water–Metal Interfaces. J. Phys. Chem. C 2020, 124, 28083–28092. [Google Scholar] [CrossRef]
- Bao, F.; Kemppainen, E.; Dorbandt, I.; Bors, R.; Xi, F.; Schlatmann, R.; Van De Krol, R.; Calnan, S. Understanding the Hydrogen Evolution Reaction Kinetics of Electrodeposited Nickel-Molybdenum in Acidic, Near-Neutral, and Alkaline Conditions. ChemElectroChem 2021, 8, 195–208. [Google Scholar] [CrossRef]
- Deluccia, J.J. Electrochemical aspects of hydrogen in metals. In Hydrogen Embrittlement: Prevention and Control; ASTM International. Special Technical Paper 962; ASTM: Philadelphia, PA, USA, 2008; pp. 17–34. [Google Scholar] [CrossRef]
- Bockris, J.O.; Reddy, A.K.; Gamboa-Aldeco, M.E. Modern Electrochemistry 2B: Fundamentals of Electrodics; Springer: Berlin/Heidelberg, Germany, 1998. [Google Scholar]
- Newman, J.; Thomas-Alyea, K.E. Electrochemical Systems, 3rd ed.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Bazant, M.Z.; Thornton, K.; Ajdari, A. Diffuse-charge dynamics in electrochemical systems. Phys. Rev. E 2004, 70, 021506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biesheuvel, P.; Bazant, M. Nonlinear dynamics of capacitive charging and desalination by porous electrodes. Phys. Rev. E 2010, 81, 031502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debye, P.; Hückel, E. The Theory of Electrolytes. 1. Freezing Point Depression and Related Phenomena. Translated from German by M. J. Braus (2020). Phys. Z. 1923, 24, 185–206. [Google Scholar]
- McCleskey, R.B.; Nordstrom, D.K.; Ryan, J.N. Comparison of Electrical Conductivity Calculation Methods for Natural Waters. Limnol. Oceanogr. Methods 2012, 10, 952–967. [Google Scholar] [CrossRef]
- Pivonka, P.; Smith, D.; Gardiner, B. Modeling of Donnan Equilibrium in Charged Porous Materials—A Scale Transition Analysis Transport. Porous Media 2007, 69, 215–237. [Google Scholar] [CrossRef]
- Biesheuvel, P.M.; Fu, Y.; Bazant, M.Z. Diffuse charge and Faradaic reactions in porous electrodes. Phys. Rev. E 2011, 83, 061507. [Google Scholar] [CrossRef] [Green Version]
- Galama, A.H.; Post, J.W.; Hamelers, H.V.M.; Nokonenko, V.V.; Biesheuvel, P.M. On the Origin of the Membrane Potential Arising Across Densely Charged Ion Exchange Membranes: How Well Does the Teorell-Meyer-Sievers Theory Work? J. Membr. Sci. Res. 2016, 2, 128–140. [Google Scholar]
- Biesheuvel, P.; Zhao, R.; Porada, S.; van der Wal, A. Theory of membrane capacitive deionization including the effect of the electrode pore space. J. Colloid Interface Sci. 2011, 360, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishchuk, N.A. Concentration polarization of interface and non-linear electrokinetic phenomena. Adv. Colloid Interface Sci. 2010, 160, 16–39. [Google Scholar] [CrossRef] [PubMed]
- Rommerskirchen, A.; Ohs, B.; Hepp, K.A.; Femmer, R.; Wessling, M. Modeling continuous flow-electrode capacitive deionization processes with ion-exchange membranes. J. Membr. Sci. 2018, 546, 188–196. [Google Scholar] [CrossRef]
- Davies, C.W. Ion Association; Butterworths: London, UK, 1962; pp. 37–53. [Google Scholar]
- Soetsbergen, M.v. Frumkin-Butler-Volmer Theory and Mass Transfer. Electrochem. Cells Russ. J. Electro-Chem. 2012, 48, 570–579. [Google Scholar]
- Biesheuvel, P.M.; Fu, Y.; Bazant, M.Z. Electrochemistry and capacitive charging of porous electrodes in asymmetric multicomponent electrolytes. Russ. J. Electrochem. 2012, 48, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Castellan, G.W. Physical Chemistry, 3rd ed.; The Benjamin-Cummings Publishing Company, Branch from Pearson Education; Pearson: San Francisco, CA, USA, 1983. [Google Scholar]
- Guiver, M.D.; Hoek, E.M.V.; Nikonenko, V.; Tarabara, V.V.; Zydney, A.L. Membrane Terminology. In Encyclopedia of Membrane Science and Technology; Wiley: Hoboken, NJ, USA, 2013; Volume 3, pp. 2219–2228. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shamim, N.; Binzaid, S.; Gabitto, J.F.; Attia, J.O. A Combined Chemical-Electrochemical Process to Capture CO2 and Produce Hydrogen and Electricity. Energies 2021, 14, 5807. https://doi.org/10.3390/en14185807
Shamim N, Binzaid S, Gabitto JF, Attia JO. A Combined Chemical-Electrochemical Process to Capture CO2 and Produce Hydrogen and Electricity. Energies. 2021; 14(18):5807. https://doi.org/10.3390/en14185807
Chicago/Turabian StyleShamim, Nabila, Shuza Binzaid, Jorge Federico Gabitto, and John Okyere Attia. 2021. "A Combined Chemical-Electrochemical Process to Capture CO2 and Produce Hydrogen and Electricity" Energies 14, no. 18: 5807. https://doi.org/10.3390/en14185807
APA StyleShamim, N., Binzaid, S., Gabitto, J. F., & Attia, J. O. (2021). A Combined Chemical-Electrochemical Process to Capture CO2 and Produce Hydrogen and Electricity. Energies, 14(18), 5807. https://doi.org/10.3390/en14185807