
Simulation-Assisted Determination of the Start-Up Time of a Polymer Electrolyte Fuel Cell




Abstract
:1. Introduction
2. Experimental
2.1. Materials and Equipment
2.2. Gas Analysis
2.3. Fuel Cell Conditioning
2.4. Fuel Cell Start-Up and Shut-Down Procedure
3. Modelling Approach
3.1. Numerical Domains
3.2. Model Calibration and Execution
3.3. Governing Equations
- Electrochemistry
- Cathode Particle Model
- Conservation of Current and Mass
- Water Transport and Mass Transfer
- Heat Sources
- Properties
- Cross-Over Current
4. Results and Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Nomenclature
| anode | |
| water activity,/ | |
| cathode | |
| CCM | catalyst coated membrane |
| CFD | computational fluid dynamics |
| specific heat of water, J (kg K)−1 | |
| diffusion coefficient between gas and liquid phase,/ | |
| liquid water diffusion coefficient, kg m−2 s−1 | |
| gas species mass diffusivity, m2 s−1 | |
| effective gas species mass diffusivity, m2 s−1 | |
| mass diffusivity of species and reference pressure and temperature , m2 s−1 | |
| diffusion coefficient of water content, kmol m−3 | |
| Euler’s number/constant | |
| user-specified activation energy anode/cathode, respectively, J mol−1 | |
| activation energy for the temperature correction term,/ | |
| reversible potentials anode/cathode, respectively, V | |
| ECSA | electrochemically active surface area, m2/g |
| equivalent weight, kg kmol−1 | |
| Faraday constant, (9.6485 × 107) C kmol−1 | |
| function for correlation of electro osmotic drag to membrane water content,/ | |
| GDL | gas diffusion layer |
| leakage current, A m−2 | |
| the ionic current density, A m−1 | |
| magnitude of solid/membrane phase current density, respectively, A m−1 | |
| reference exchange current density, respectively, A m−2 | |
| reference exchange current density at specified reference temperature anode/cathode, respectively, A m−2 | |
| ideal oxygen molar flux, mol m−2 s−1 | |
| absolute permeability,/ | |
| relative permeability,/ | |
| product of oxygen solubility and diffusivity in liquid water, respectively, m2 s−1 | |
| latent heat of evaporation, J kg−1 | |
| MPL | micro porous layer |
| molecular mass of hydrogen, oxygen, water vapor, ionomer, respectively, kg kmol−1 | |
| osmotic drag coefficient,/ | |
| user-specified generality coefficient for electro osmotic drag coefficient,/ | |
| NLPM | normal liters per minute |
| OCV | open circuit voltage |
| pressure, Pa | |
| capillary pressure, Pa | |
| liquid pressure gradient, kg m2 s−2 | |
| partial pressure of water vapor, Pa | |
| partial pressures of hydrogen, oxygen and water vapor, respectively, Pa | |
| water vapor saturation pressure, Pa | |
| user-specified standard state absolute pressure, Pa | |
| reference pressure, Pa | |
| PEM | proton exchange membrane |
| PEMFC | proton exchange membrane fuel cell |
| PFSA | perfluoro sulfonic acid |
| PTFE | polytetrafluoroethylene |
| universal gas constant, (8.3144) J (mol K)−1 | |
| exchange current density anode/cathode, respectively, A m−3 | |
| transfer current modification due to occurrence of liquid water, A m−3 | |
| particle diameter, m | |
| exponent of pore blockage,/ | |
| ideal transfer current, A m−2 s−1 | |
| volumetric transfer current, respectively, A m−3 | |
| resistance due to ionomer film/liquid water film surrounding particles, respectively, s m−1 | |
| RH | relative humidity, % |
| liquid saturation,/ | |
| volumetric heat source for the gas diffusion layer and micro porous layer, anode catalyst layer, membrane and current collector, respectively, J m−3 s−1 | |
| sink/source terms for hydrogen, oxygen, membrane water content, between gas and dissolved phases, liquid and dissolved phases and gas and liquid phases, respectively, kg m−3 s−1 | |
| reaction entropies anode/cathode, respectively, J K−1 | |
| temperature, K | |
| user-specified reference temperature anode/cathode, respectively,/ | |
| user-specified standard state temperature, K | |
| reference temperature, K | |
| half cell potential anode/cathode, respectively, V | |
| volume, m3 | |
| gas velocity vector, m s−1 | |
| liquid water velocity vector, m s−1 | |
| volume of the anode/cathode catalyst layer, m3 | |
| local species concentration, kmol m−3 | |
| Greek alphabet | |
| anode and cathode transfer coefficients of the anode electrode, respectively,/ | |
| anode and cathode transfer coefficients of the cathode electrode, respectively,/ | |
| electrolyte phase conductivity model generality constant in membrane, anode catalyst and cathode catalyst, respectively,/ | |
| electrolyte phase conductivity term,/ | |
| concentration dependence coefficient anode/cathode, respectively,/ | |
| evaporation/condensation coefficient, respectively,/ | |
| user-specified constant for modification of transfer current due to occurrence of liquid water,/ | |
| user-specified parameters for mass exchange rate between gas and dissolved/liquid and dissolved phases,/ | |
| user-defined exponents in gas species diffusivity equation for temperature/pressure, respectively,/ | |
| porosity,/ | |
| porosity of porous media,/ | |
| specific active area anode/cathode (cathode catalyst), respectively, m−1 | |
| surface over potential anode/cathode, V | |
| coefficient of liquid water removal,/ | |
| user-specified parameter,/ | |
| contact angle, ° | |
| membrane water content, dissolved water content,/ | |
| equilibrium water content,/ | |
| water activity at water activity equal to 1, saturation equal to 1, i.e., user-specified parameters,/ | |
| liquid dynamic (absolute) viscosity, kg (ms)−1 | |
| dry ionomer, i.e., membrane, density, kg m−3 | |
| liquid water density, kg m−3 | |
| surface tension coefficient, N m−1 | |
| electrical conductivity of the solid/membrane phase, respectively, (Ωm)−1 | |
| ionomer volume fraction anode/cathode, respectively,/ | |
| ionomer tortuosity anode/cathode,/ | |
| electric potential of the solid/membrane phase, respectively, V | |
| liquid to gas velocity ratio,/ | |
| electrolyte phase conductivity model generality constant,/ | |
| Mathematical operator | |
| nabla operator | |
References
- Oyarce, A.; Zakrisson, E.; Ivity, M.; Lagergren, C.; Ofstad, A.B.; Bodén, A.; Lindbergh, G. Comparing shut-down strategies for proton exchange membrane fuel cells. J. Power Sources 2014, 254, 232–240. [Google Scholar] [CrossRef]
- Lin, R.; Liu, D.; Xia, S.; Ma, T.; Dutruel, B. Stack shut-down strategy optimisation of proton exchange membrane fuel cell with the segment stack technology. Int. J. Hydrogen Energy 2019, 45, 1030–1044. [Google Scholar] [CrossRef]
- Kim, J.H.; Cho, E.A.; Jang, J.H.; Kim, H.J.; Lim, T.H.; Oh, I.H.; Ko, J.J.; Son, I.-J. Development of a Durable PEMFC Start-Up Process by Applying a Dummy Load. J. Electrochem. Soc. 2010, 157, B118–B124. [Google Scholar] [CrossRef]
- Bodner, M.; Cermenek, B.; Rami, M.; Hacker, V. The Effect of Platinum Electrocatalyst on Membrane Degradation in Polymer Electrolyte Fuel Cells. Membranes 2015, 5, 888–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coms, F.D. The Chemistry of Fuel Cell Membrane Chemical Degradation Frank D. Coms General Motors Corporation, Fuel Cell Research Labs, 10 Carriage Street, Honeoye Falls, NY 14472, USA. ECS Trans. 2008, 16, 235–255. [Google Scholar] [CrossRef]
- Kusoglu, A.; Weber, A.Z. New Insights into Perfluorinated Sulfonic-Acid Ionomers. Chem. Rev. 2017, 117, 987–1104. [Google Scholar] [CrossRef]
- Bodner, M.; Schenk, A.; Salaberger, D.; Rami, M.; Hochenauer, C.; Hacker, V. Air Starvation Induced Degradation in Polymer Electrolyte Fuel Cells. Fuel Cells 2017, 17, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wang, P.; Chen, H.; Pei, P. A review of automotive proton exchange membrane fuel cell degradation under start-stop operating condition. Appl. Energy 2018, 223, 249–262. [Google Scholar] [CrossRef]
- Bodner, M.; Senn, J.; Hacker, V. Degradation Mechanisms and Their Lifetime; Elsevier: Amsterdam, The Netherlands, 2018; pp. 139–154. [Google Scholar] [CrossRef]
- Iranzo, A.; Boillat, P. CFD simulation of the transient gas transport in a PEM fuel cell cathode during AC impedance testing considering liquid water effects. Energy 2018, 158, 449–457. [Google Scholar] [CrossRef]
- Dadda, B.; Abboudi, S.; Ghezal, A. Transient two-dimensional model of heat and mass transfer in a PEM fuel cell membrane. Int. J. Hydrogen Energy 2013, 38, 7092–7101. [Google Scholar] [CrossRef]
- Peksen, M. 3D transient multiphysics modelling of a complete high temperature fuel cell system using coupled CFD and FEM. Int. J. Hydrogen Energy 2014, 39, 5137–5147. [Google Scholar] [CrossRef]
- Shimpalee, S. Dynamic Simulation of Large Scale PEM Fuel Cell under Driving Cycle. J. Electrochem. Soc. 2014, 161, E3138–E3148. [Google Scholar] [CrossRef]
- Tolj, I.; Penga, Ž.; Vukičević, D.; Barbir, F. Thermal management of edge-cooled 1 kW portable proton exchange membrane fuel cell stack. Appl. Energy 2019, 257, 114038. [Google Scholar] [CrossRef]
- Penga, Ž.C.; Bergbreiter, C.; Barbir, F.; Scholta, J. Numerical and experimental analysis of liquid water distribution in PEM fuel cells. Energy Convers. Manag. 2019, 189, 167–183. [Google Scholar] [CrossRef]
- Bezmalinović, D.; Radošević, J.; Barbir, F. Initial conditioning of polymer eelectrolyte membrane fuel cell by temperature and potential cycling. Acta Chim. Slov. 2015, 62, 83–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocher, K.; Kolar, S.; Ladreiter, W.; Hacker, V. Cold start behavior and freeze characteristics of a polymer electrolyte membrane fuel cell. Fuel Cells 2021, 21, 363–372. [Google Scholar] [CrossRef]
- Scholz, H. Modellierung und Untersuchung von Flutungsphänomenen in Niedertemperatur-PEM-Brennstoffzellen. Aachen, Germany, 2015. Available online: http://publications.rwth-aachen.de/record/570114 (accessed on 18 November 2021).
- Wu, H.; Li, X.; Berg, P. On the modeling of water transport in polymer electrolyte membrane fuel cells. Electrochim. Acta 2009, 54, 6913–6927. [Google Scholar] [CrossRef]
- Zawodzinski, T.A.; DeRouin, C.; Radzinski, S.; Sherman, R.J.; Smith, V.T.; Springer, T.E.; Gottesfeld, S. Water Uptake by and Transport Through Nafion®® 117 Membranes. J. Electrochem. Soc. 1993, 140, 1041–1047. [Google Scholar] [CrossRef]
- Hinatsu, J.T.; Mizuhata, M.; Takenaka, H. Water Uptake of Perfluorosulfonic Acid Membranes from Liquid Water and Water Vapor. J. Electrochem. Soc. 1994, 141, 1493–1498. [Google Scholar] [CrossRef]
- Penga, Ž.I.; Tolj, I.; Barbir, F. Computational fluid dynamics study of PEM fuel cell performance for isothermal and non-uniform temperature boundary conditions. Int. J. Hydrogen Energy 2016, 41, 17585–17594. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.-Y. Transient analysis of polymer electrolyte fuel cells. Electrochim. Acta 2004, 50, 1307–1315. [Google Scholar] [CrossRef]
- Motupally, S.; Becker, A.J.; Weidner, J. Diffusion of Water in Nafion 115 Membranes. J. Electrochem. Soc. 2000, 147, 3171–3177. [Google Scholar] [CrossRef]
- Springer, T.E.; Zawodzinski, T.A.; Gottesfeld, S. Polymer Electrolyte Fuel Cell Model. J. Electrochem. Soc. 1991, 138, 2334–2342. [Google Scholar] [CrossRef]




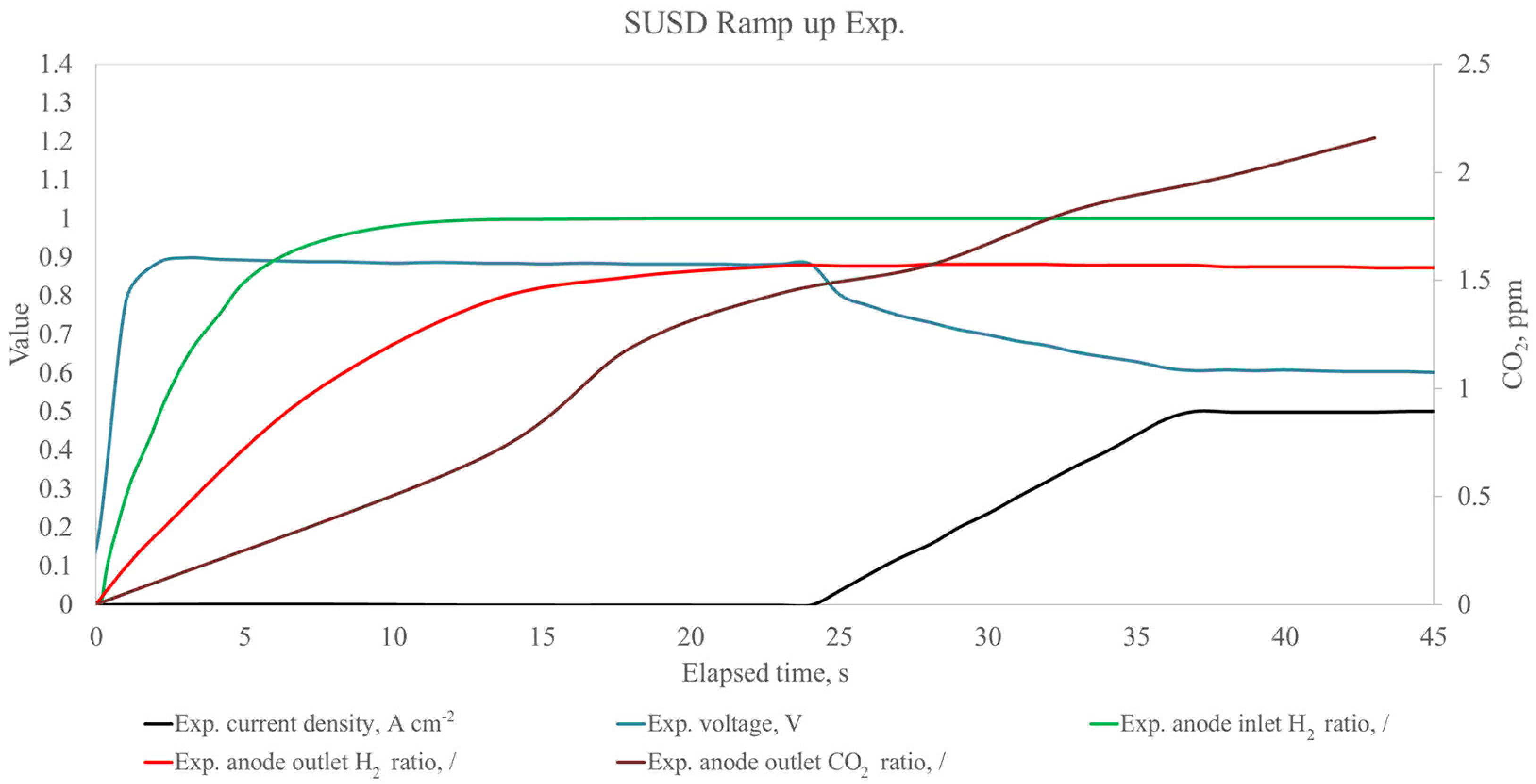





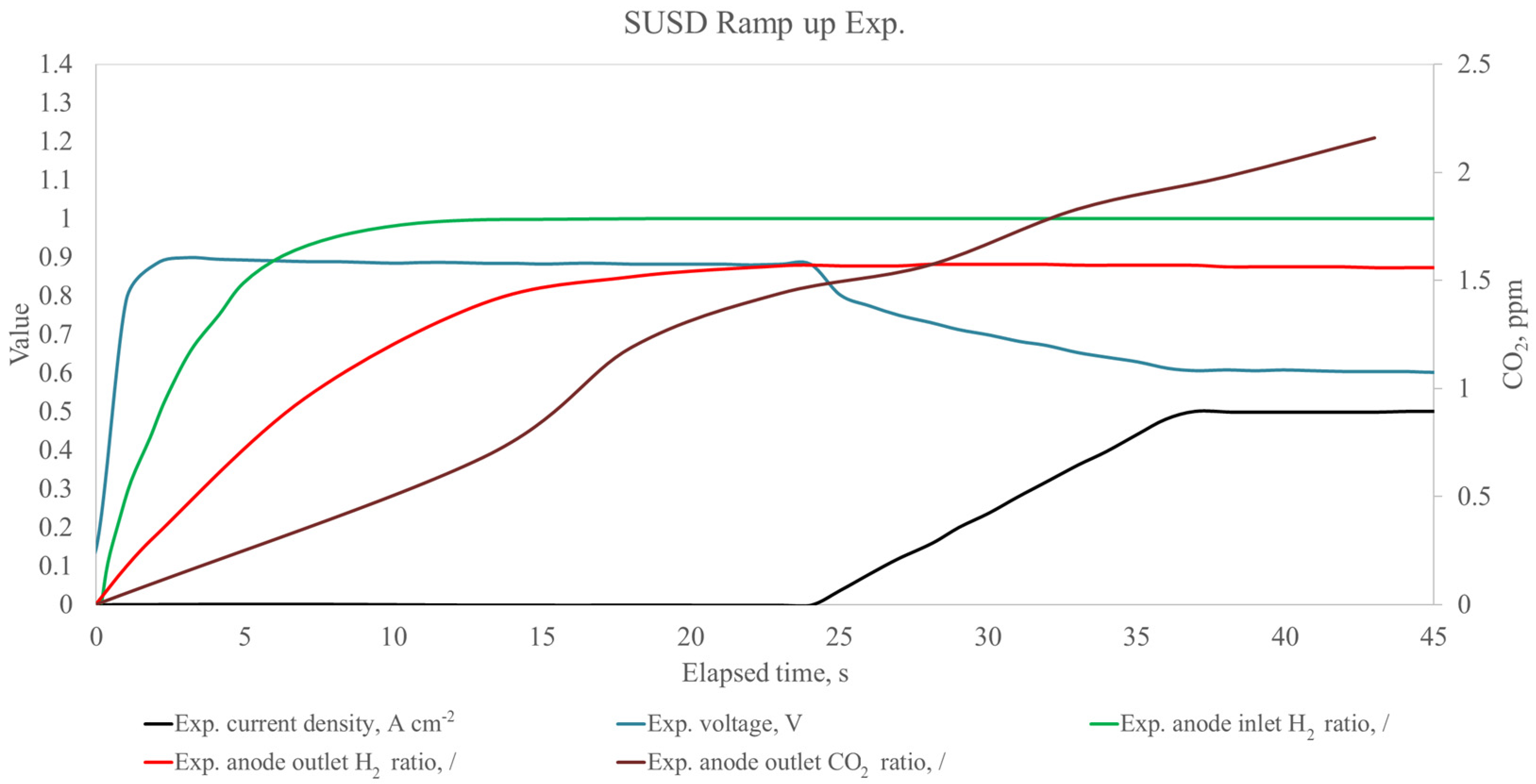
{kind=link}
{kind=link}
{kind=link}
{kind=link}
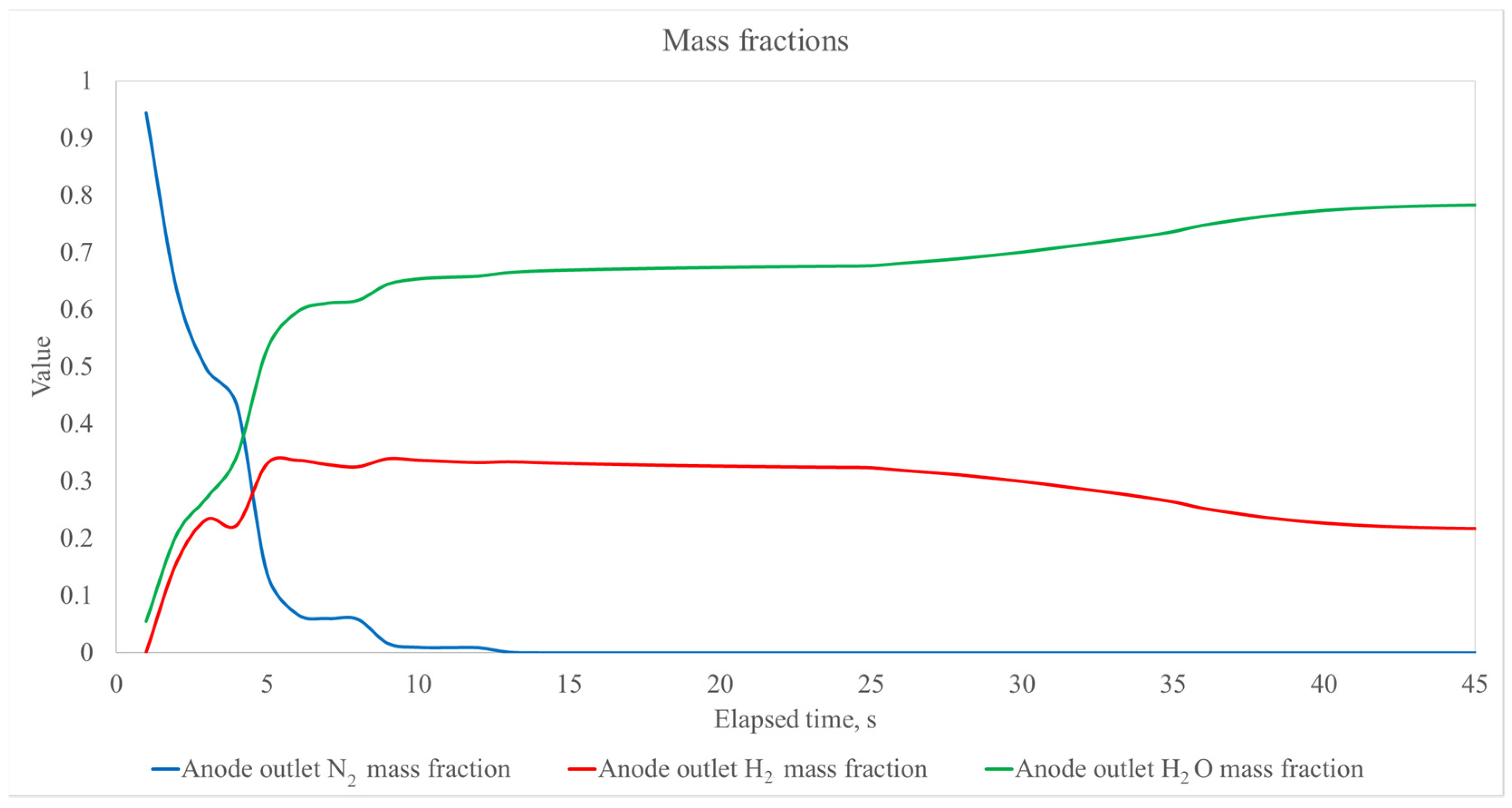
{kind=link}
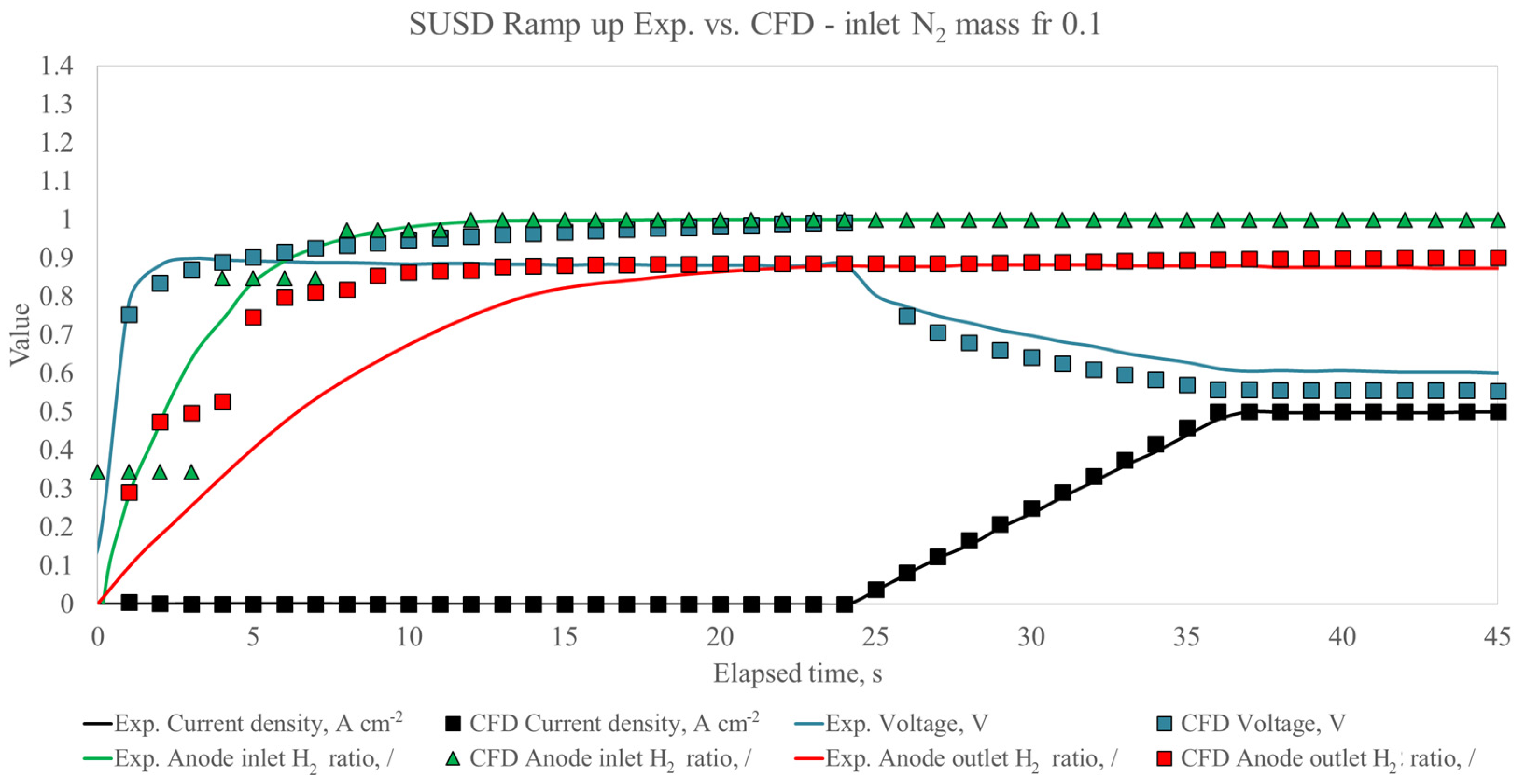{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit | Anode | Cathode | |
|---|---|---|---|
| p | kPag | 100 | 100 |
| RH | 80% | 60% | |
| Tcell | °C | 80 | 80 |
| Tdp | °C | 75 | 68 |
| Tinlet | °C | 85 | 85 |
| gas flow rate | NLPM | 0.4 | 0.4 |
| Layer | Thickness, µm |
|---|---|
| Gas diffusion substrate | 155 |
| MPL | 20 |
| Catalyst | 10 |
| Membrane | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodner, M.; Penga, Ž.; Ladreiter, W.; Heidinger, M.; Hacker, V. Simulation-Assisted Determination of the Start-Up Time of a Polymer Electrolyte Fuel Cell. Energies 2021, 14, 7929. https://doi.org/10.3390/en14237929
Bodner M, Penga Ž, Ladreiter W, Heidinger M, Hacker V. Simulation-Assisted Determination of the Start-Up Time of a Polymer Electrolyte Fuel Cell. Energies. 2021; 14(23):7929. https://doi.org/10.3390/en14237929
Chicago/Turabian StyleBodner, Merit, Željko Penga, Walter Ladreiter, Mathias Heidinger, and Viktor Hacker. 2021. "Simulation-Assisted Determination of the Start-Up Time of a Polymer Electrolyte Fuel Cell" Energies 14, no. 23: 7929. https://doi.org/10.3390/en14237929
APA StyleBodner, M., Penga, Ž., Ladreiter, W., Heidinger, M., & Hacker, V. (2021). Simulation-Assisted Determination of the Start-Up Time of a Polymer Electrolyte Fuel Cell. Energies, 14(23), 7929. https://doi.org/10.3390/en14237929