Abstract
Understanding the processes occurring during the nanocrystallization of glass-forming liquids is important for creating artificial nanostructures for various applications. In this article, local thermal perturbations in supercooled glass-forming liquids and polymers during the nucleation of a crystalline phase are studied. To describe the thermal response of supercooled glass-forming liquids, an integro-differential heat equation with dynamic heat capacity is used. We have found that the effect of the dynamic heat capacity is significant for fast local thermal perturbations that arise in the early stages of crystal nucleation in glass-forming liquids and polymers. It has been established that local temperature perturbations during the nucleation of crystals in silicate glasses and polymers can change the nucleation rate by 2–5 orders of magnitude. The knowledge gained can be useful for the technology of artificial microstructures and advanced materials.
1. Introduction
This work is devoted to the problem of heat transfer, which is associated with nonequilibrium thermal processes in metastable liquids. In this study, we are interested in nonequilibrium heat transfer in undercooled glass-forming liquids, in which the process of the early stages of crystal nucleation occurs at the nanoscale. Glasses and polymers are often used to develop new advanced nanocrystalline materials and composites. The properties of glass-forming materials can be varied over a wide range by controlling the crystallization process with appropriate heat treatments, without changing the composition. Glass-forming materials are materials that form glasses at relatively moderate cooling rates (i.e., do not require extreme quenching). Accordingly, the glass-forming liquid is a liquid melt of the glass-forming material. Various artificial nanostructures can be created using the controlled crystallization of glass-forming liquids; this fact is used in many applications, such as additive manufacturing [1,2,3,4]. The fields of application of glass-forming materials can be greatly extended using optimal annealing treatments [5]. Thus, by the appropriate annealing of glass-forming materials, a mixed glassy-nanocrystalline structure can be obtained [5,6]. Nanosized crystalline particles, selectively introduced into the glass matrix, can significantly improve the mechanical properties of glasses. For example, the mechanical properties of aluminum-based glasses have been significantly improved by selectively introducing nanosized crystalline particles into the glass matrix [7,8,9]. In addition, an artificially created microstructure can be used to increase the kinetic stability of such nanoglasses [5,6]. In addition, understanding the behavior of subcritical nuclei is important for developing advanced materials for phase-change memory devices [10]. Indeed, the role of the crystallization process is very important in laser memory devices with a melting–crystallization phase transition. A phase-change material should have as fine a nanostructure as possible. In addition, it should neither be too difficult to crystallize (because it would require excessive power or conversion time) nor crystallize too easily (because it would compromise long-term data storage by initiating unwanted crystallization) [10]. In fact, subcritical nuclei are constantly developing and decaying. Only near-critical nuclei can commonly reach the critical size and grow into stable crystal grains (note that a small local change in temperature can significantly affect the dynamics of near-critical nuclei). Experimental studies of the early stages of crystallization are challenging due to the nanometer size and the very short lifetime of subcritical and near-critical nuclei. However, the nucleation of crystals can often be studied at low supercooling, and at very high supercooling, near the glass transition temperature, and in the case of liquids with low crystal-growth rates, even over the entire supercooling range from the melting temperature to the glass transition temperature [11]. Thus, developing new promising nanocrystalline glass-forming materials requires a deep understanding of the kinetics of the early stages of crystal nucleation. In fact, the formation of nanocrystallized materials is commonly associated with homogeneous nucleation [12,13,14,15]. In this paper, we will focus on homogeneous nucleation in glass-forming liquids above the glass transition temperature, .
Local temperature changes associated with the energetics of nuclei formation can significantly affect nucleation dynamics. In this article, we study the influence of the local temperature perturbations associated with the nucleation of crystals on the processes of the early stage of nucleation in glass-forming liquids. We will not consider crystal precursors in undercooled liquids [16,17]. Thus, we consider homogeneous nucleation which is not affected by pre-existing clusters. The scenario of two-stage nucleation, which can occur due to the formation of precursors, can be considered in a separate article. Here, our goal is to study the effect of local temperature perturbations associated with crystal nucleation on the formation of nuclei in glass-forming liquids, and to study heat transfer processes at the stage of crystal nucleation. Note also that the local temperature distribution was considered for the non-isothermal homogeneous nucleation of water vapor under quasi-isothermal conditions in [18]. Here, we are interested in the local temperature change during nucleation in condensed matter. In addition, we consider the effect of dynamic heat capacity relaxation on the heat transfer at this stage of nucleation.
The dynamic heat capacity of glass-forming liquids and polymers as a function of time (or frequency) has been intensively studied since the pioneering work of Birge and Nagel [19]. For example, see heat capacity spectroscopy in polymers [20,21]. The time dependence is associated with slow energy exchange between different degrees of freedom in glass-forming liquids and polymers. Because is time-dependent, the local change in temperature over time depends on the temperature distribution in these liquids at earlier times. This behavior can be described using the integro-differential heat equation with “memory” [22]. The influence of the dynamic heat capacity is most significant for fast local thermal perturbations on the nanometer scale [22]. This work aims to determine the local thermal perturbations (associated with the nucleation of crystals) and the effect of these perturbations on the nucleation rate in glass-forming liquids.
The first part of the article considers the influence of a local temperature change on the nucleation rate in silicate glasses and polymers. In the second part, the influence of the dynamic heat capacity on thermal perturbations in glass-forming liquids is estimated. An analytical solution of the integro-differential heat equation with dynamic heat capacity is constructed for a spherically symmetric problem. In fact, we consider a simple model of creation and decay of a spherical nucleus under spherically symmetric boundary conditions. Finally, local temperature perturbations during the nucleation and decay of crystalline nuclei in silicate glasses and polymers are considered.
2. Crystal Nucleation in Glass-Forming Liquids
Isothermal processes are considered in the classical theory of homogeneous nucleation [23,24,25,26,27]. This consideration is justified for materials with moderate and high thermal conductivity. However, glass-forming liquids, especially polymers, can have a low thermal conductivity of the order of 0.1 W/m·K [28]. For this reason, local temperature changes associated with the nucleation process can be significant on the nanosecond time scale (see below). In addition, the nonequilibrium thermal response of glasses and polymers is associated with the relaxation of the dynamic heat capacity, which can significantly affect local thermal perturbations in these substances during crystal nucleation [22,29]. Note also that the pre-exponential factor of the rate of homogeneous nucleation, measured in many experiments, significantly exceeds the theoretical value [12,13,30,31]. The discrepancy between the experimental and theoretical values of the pre-exponential factor can possibly be reduced by taking into account the temperature dependence of the interfacial free energy [13] and seeded crystallization [31,32]. In this article, we explore an additional cause that can affect the rate of nucleation.
The probability of a thermodynamic fluctuation creating a crystal nucleus is proportional to , where is Boltzmann’s constant, and is the minimum work required to create a nucleus [23,24]. A local pressure perturbation associated with a thermodynamic fluctuation of radius relaxes on a time scale of the order of , where the speed of sound is about several thousand m/s. Thus, the local pressure relaxes on a picosecond scale at of the order of several nm or fewer. Therefore, the local pressure near such fluctuations can be considered constant on a nanosecond time scale. The time required for the formation of a crystalline nucleus from the beginning of its formation is denoted by . This formation time should not be confused with the induction time and the time lag before the beginning of the formation of crystal nuclei (for example, in molecular dynamics simulations). Usually, the time lag before the beginning of the formation of crystal nuclei is much longer than [33]. In fact, the formation of crystal nuclei with a size of the order of 1 nm in various materials can occur over a time in the range of 0.01–1 ns [4,33,34,35,36,37,38], and in polymers, can vary on the nanosecond scale [39,40,41,42,43].
For a spherical nucleus, is equal to the Gibbs free energy
required for the creation of a nucleus of radius , where is the solid/liquid interfacial energy, is the change in volumetric Gibbs free energy at crystallization, is the volumetric enthalpy of melting, is the equilibrium melting temperature, and is undercooling [12,23,24,25,26,27,44]. Furthermore, we will consider critical and subcritical nuclei with , where the critical radius corresponds to the energy barrier for homogeneous nucleation , or [12,23,24,25,26,27]. Therefore, , where is the volume of the critical nucleus.
In the case of polymers, the Gibbs free energy required for the formation of a lamellar nucleus with thickness and radius is equal to
where and correspond to the surface free energy of the lamellar basal plane (fold surface) and the lateral (side) interface, respectively [13,16]. The approximate free-energy difference between the liquid and crystal can be corrected and presented as (which is often used for polymers [13]), or even [44]. However, these corrections are not significant for the present study, since our estimates do not claim to be highly accurate. Thus, we will consider the following approximation: . In this case, the critical radius and critical thickness are associated with the energy barrier for homogeneous nucleation [13,16]. Therefore, , where is the volume of the critical nucleus.
The nucleus of the solid phase, created due to thermodynamic fluctuations, has additional energy compared to the metastable initial liquid phase. The energy spent on the creation of the nucleus is compensated by the local cooling of the material at the site of the nucleus. This local change in temperature , associated with the energy required for the formation of nuclei, can significantly affect the dynamics of the nucleation and evolution of nuclei. In fact, a significant number of unstable nuclei (with ) can be transformed into stable nuclei (with ), since the critical radius decreases with decreasing the local temperature . We will focus on local fast thermal perturbations in glass-forming liquids above the glass transition temperature . Therefore, we will not consider the effect of long-term structural relaxation, which is noticeable near and below , on the nucleation dynamics [45,46,47,48]. However, we consider the effect associated with the dynamic heat capacity, which is significant for fast local thermal perturbations in glass-forming liquids, even well above the glass transition temperature [22,29].
2.1. Influence of Local Temperature Change on the Nucleation Rate in Silicate Glasses
The local change in temperature associated with the energy required for the formation of nuclei can be about 1 K (see below). In this case, the evolution of near-critical nuclei and the nucleation rate can change significantly. In fact, the nucleation rate is proportional to [12,13,23,24,25,26,27]. The relative increase in the rate is equal to
at (an insignificant value of compared to is neglected). Table 1 shows the influence of the thermal perturbation on the relative change in the rate of nucleation at −1 K for silicate glasses. The parameters of the silicate glasses are known from the literature [12,49,50,51,52,53].

Table 1.
Relative increase in the nucleation rate in silicate glasses at −1 K.
Next, we estimate the effect of the kinetic barrier on the nucleation rate under thermal perturbations in silicate glasses. In fact,
where is the kinetic barrier to nucleation [49,50]. The kinetic barrier can be estimated from the activation free energy for a viscous flow [49,50]. Then,
since the viscosity usually obeys the Vogel–Fulcher–Tammann–Hesse (VFTH) equation [52,53].
Thus,
where
Table 2 shows the influence of the thermal perturbation on the relative change in the rate of nucleation due to a change in the kinetic barrier at −1 K for silicate glasses. The parameters of the silicate glasses are known from the literature [52,53].
Table 2.
Relative change in the nucleation rate due to a change in the kinetic barrier in silicate glasses at −1 K.
Thus, the effect of the kinetic barrier on the nucleation rate at small temperature perturbations in silicate glasses is insignificant. However, thermal perturbations of the order of 1 K change the nucleation rate by 2–4 orders of magnitude (see Table 1), which is significant.
2.2. Influence of Local Temperature Change on the Nucleation Rate in Polymers
The relative change in the rate of nucleation for glass-forming polymers is equal to at (see Equation (3)), where . As mentioned above, the small value of compared to is neglected. Table 3 and Table 4 show the influence of the thermal perturbation on the relative change in the rate of nucleation for polyethylene (PE) and isotactic polystyrene (iPS) at −0.5 K (see Table 3), as well as polybutylene succinate (PBS) and isotactic polypropylene (iPP) at −1 K (see Table 4). Temperature perturbations −0.5 K (for PE and iPS) and −1 K (for PBS and iPP) were evaluated as described below. The parameters of the polymers are known from the literature [13,54,55,56,57,58,59,60].
Table 3.
Relative increase in the nucleation rate in PE and iPS at −0.5 K.
Table 4.
Relative increase in the nucleation rate in PBS and iPP at −1 K.
The influence of the kinetic barrier on the nucleation rate under thermal perturbations in polymers can be estimated as follows. The relative change in the rate of nucleation is equal to (see Equation (7)), where is equal to (see Equation (8)), and depends on the diffusional activation energy for the transport of polymer chain segments at the interface in polymers [13]; 8.314 J/mol·K is the universal gas constant [28], and usually, 6.27 kJ/mol and [13]. The effect of the kinetic barrier on the nucleation rate under thermal perturbations in polymers is presented in Table 5 and Table 6. The parameters of the polymers are known from the literature [13,54,55,56,57,58].
Table 5.
Relative change in the nucleation rate due to a change in the kinetic barrier in PE and iPS at −0.5 K.
Table 6.
Relative change in the nucleation rate due to a change in the kinetic barrier in PBS and iPP at −1 K.
3. Thermal Perturbations in Glass-Forming Liquids
Next, we consider heat transfer processes in glass-forming liquids in order to determine the magnitude of thermal perturbations during the nucleation and decay of crystal nuclei. Since we are interested in thermal perturbations on a nanosecond time scale, it is necessary to take into account the influence of the dynamic heat capacity of glass-forming liquids. For a spherically symmetric problem with zero initial conditions, the thermal response in glass-forming liquids can be described by the following integro-differential heat equation [22,29]:
where and are the density and thermal conductivity of the material; , —Laplacian, and are the volumetric heat flux density. Equation (9) has an analytical solution for a homogeneous boundary value problem. Assume that the dynamic heat capacity is a completely monotonic function of time (this requirement is usually satisfied for glass-forming liquids [22]). Then, can be represented by Equation (10)
where is the distribution function of the relaxation time ; a and are the initial and equilibrium heat capacities ( as , and as ). To determine for a particular material, broadband heat capacity spectroscopy can be used (for details, see [22]).
The solution of the boundary value problem for Equation (9) can be obtained in two stages. First, the problem is solved for the heat capacity with Debye relaxation, shown in Equation (11),
where . After that, using the distribution function , the solution for a particular material can be obtained as a linear combination of solutions for different . Thus, Equation (12) follows on from Equations (9) and (11) for the heat capacity with Debye relaxation:
where is the equilibrium thermal diffusivity. The parameter of various glass-forming liquids and polymers is usually in the range of 0.3–0.5 [53,54,55,56,61,62,63]. First, assume that a spherically symmetric heat source is distributed in a spherical volume of radius . Next, consider the homogeneous boundary condition at a distance from the center of the heat source and the initial condition for . Then, Equation (13) is a solution to this boundary value problem.
where must satisfy Equation (14):
where and . Here, let the heat flux be uniformly distributed in the volume of radius with volumetric density . Then,
For example, consider a sinusoidal heat flux with for and for . Then, as follows from Equation (14), are represented by Equation (16) and Equation (17) for and , respectively.
where and are equal to the roots of the polynomial . In addition, and are real and positive parameters; for and , .
4. Local Thermal Perturbations during the Nucleation and Decay of Crystal Nuclei in Glass-Forming Liquids
To estimate the magnitude of the local temperature change δT(t,r) during the nucleation in glass-forming liquids, we consider the nucleation of spherical crystalline nuclei. For a boundary value problem at , the size of is not significant. For example, we take 300 nm. Next, we consider the nucleation and decay of a nucleus with a subcritical radius of the order of 1 nm during 1 ns. The sinusoidal heat flux density can be considered as a simple model for the formation and decay of crystalline nuclei. The heat flux density required to compensate for the costs of nucleation is equal to . Therefore, , since .
Consider, for example, the silicate glass Na2O·2CaO·3SiO2 with the thermal parameters given in Table 1. Thus, we take 200 K and use the parameters of this glass in the liquid phase [53,62,63,64,65]. Thus, the following parameters were used for model calculations: 2.8 g/cm3, 5 × 106 J/m3K, 0.5 W/m·K, 1 × 10−7 m2/s, , 1560 K, 7.2 × 108 J/m3, and = 292 nm3. Let us consider nuclei close to the critical size. Then, at , we have = 4.1 nm. Then, can be calculated according to Equation (13) at various . For example, 0, 0.1 ns, 1 ns, 10 ns, and 100 ns (see Figure 1 and Figure 2). Note also that the sum of series (13) converges with the sum of series . Therefore, to obtain a result with an accuracy of about 1%, it is enough to calculate the sum up to terms, since the remainder of the sum is about .
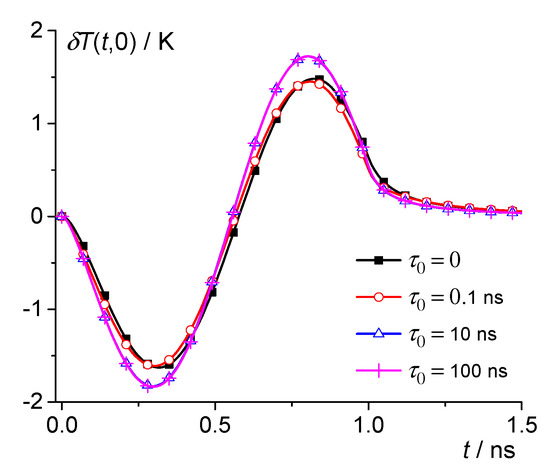
Figure 1.
Dependence of the temperature change on time for the silicate glass Na2O·2CaO·3SiO2 at 200 K; 0, 0.1 ns, 10 ns, and 100 ns—squares, circles, triangles, and crosses.
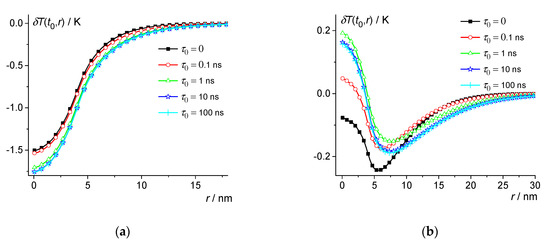
Figure 2.
Dependence of the temperature change on distance at 0.25 ns (a) and 0.57 ns (b) for the silicate glass Na2O·2CaO·3SiO2 at 200 K; 0, 0.1 ns, 1 ns, 10 ns, and 100 ns—squares, circles, triangles, stars, and crosses.
Thus, the magnitude of the local temperature change δT(t,r) during the nucleation and decay of subcritical nuclei can reach about 1 K in silicate glass-forming liquids. The effect of dynamic heat capacity is significant at 10 ns (see Figure 1 and Figure 2). However, at 0.1 ns, this effect is small. The effect increases with and reaches saturation when approaches 10 ns (see Figure 2).
Similar results can be obtained for polymers (see Figure 3, Figure 4, Figure 5 and Figure 6). Consider, for example, isotactic polystyrene (iPS) with the thermal parameters given in Table 3. Thus, we take 30 K and use the parameters of iPS in the liquid phase [13,54,55,56]. Thus, the following parameters were used for model calculations: 1.05 g/cm3, 1.8 × 106 J/m3K, 0.14 W/m·K, 0.8 × 10−7 m2/s, , 516 K, 0.91 × 108 J/m3, and = 704.5 nm3. Let us consider nuclei close to the critical size. Then, at , we have = 5.5 nm. For example, we calculate using Equation (13) at 0, 0.1 ns, 1 ns, 10 ns, and 100 ns, as well as when averaged over the distribution (see Figure 3 and Figure 4). The distribution can be obtained from the broad band heat capacity spectroscopy (for details, see [22]). Moreover, relaxation phenomena in glass-forming materials can usually be described by the Kohlrausch relaxation function , where is about 0.5. In addition,
at 0.5 [66]. The Kohlrausch relaxation time can be obtained from the Vogel–Fulcher–Tammann–Hesse (VFTH) equation
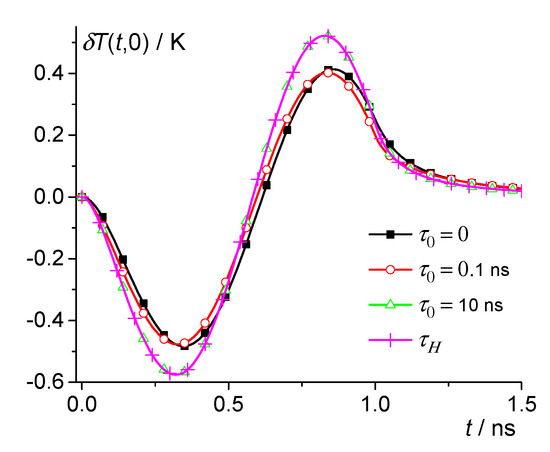
Figure 3.
Dependence of the temperature change on time for iPS at 30 K. 0, 0.1 ns, and 10 ns—squares, circles, and triangles; crosses are the result of averaging over the distribution .
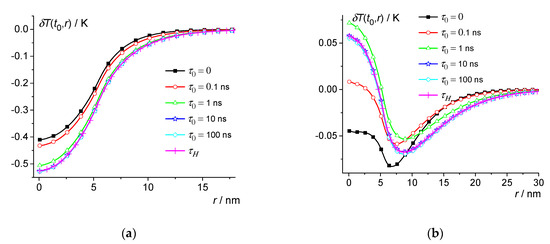
Figure 4.
Dependence of the temperature change on distance at 0.25 ns (a) and 0.6 ns (b) for iPS at 30 K. 0, 0.1 ns, 1 ns, 10 ns, and 100 ns—squares, circles, triangles, stars, and diamonds; crosses are the result of averaging over the distribution .
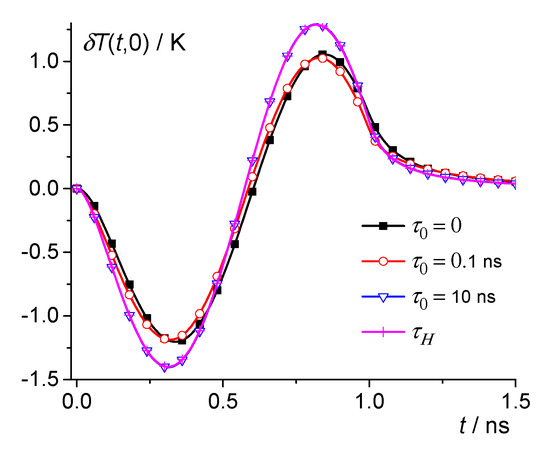
Figure 5.
Dependence of the temperature change on time for PBS at 60 K. 0, 0.1 ns, and 10 ns—squares, circles, and triangles; crosses are the result of averaging over the distribution .
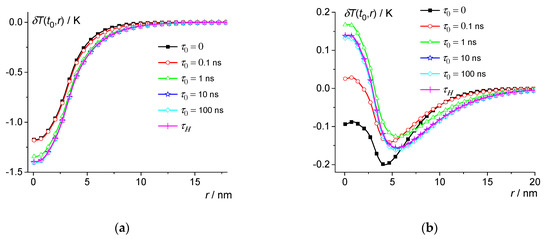
Figure 6.
Dependence of the temperature change on distance at 0.3 ns (a) and 0.59 ns (b) for PBS at 60 K. 0, 0.1 ns, 1 ns, 10 ns and 100 ns—squares, circles, triangles, stars, and diamonds; crosses are the result of averaging over the distribution .
The parameters of this equation can be measured using broad band heat capacity spectroscopy [21]. In fact, is inversely proportional to the frequency , which corresponds to the maximum of the imaginary part of the dynamic heat capacity (see [22]). For polystyrene, = 10.2, = 388 K, and = 341.5 K [21]. Thus, we obtain the Kohlrausch relaxation time and the distribution function at different temperatures from the VFTH equation. However, the profile of the distribution function is not very important, because the influence of the dynamic heat capacity reaches saturation at of the order of tens of ns (see Figure 4).
Thus, the magnitude of the local temperature change δT(t,r) during the nucleation in iPS is about 0.5 K (a similar result was obtained for PE). The effect of dynamic heat capacity reaches saturation at 10 ns (see Figure 3 and Figure 4). As a consequence, at 10 ns and 100 ns are practically the same and coincide with the result of averaging over the distribution (see Figure 3 and Figure 4). Thus, the effect of dynamic heat capacity is significant on the nanosecond time scale. It should be noted that due to the dynamic heat capacity, thermal perturbations at 0.1 ns and 0 even have opposite signs at 0.6 ns and 5 nm (see Figure 4b).
A larger local temperature change δT(t,r) can be estimated for the biodegradable polymer polybutylene succinate (PBS) (see Table 4). For example, we take 60 K and use the parameters of polybutylene succinate in the liquid phase [54,55,56,57,58,59,60]. For model calculations, we use the following parameters: 1.26 g/cm3, 2.4 × 106 J/m3K, 0.09 W/m·K, 0.4 × 10−7 m2/s, , 403 K, 1.47 × 108 J/m3, and = 152.6 nm3, and consider nuclei close to the critical size. Then, at , we have = 3.3 nm. The distribution function for PBS at different temperatures can be obtained from dielectric spectroscopy data using the parameters ( 9.5, 526.3 K, and 218.2 K) of the VFTH equation [67].
The magnitude of the local temperature change during the nucleation in PBS is about 1 K (see Figure 5 and Figure 6) (a similar result was obtained for iPP). The effect of dynamic heat capacity reaches saturation when increases above tens of ns. As a result, the profile of the distribution function is not very important (see Figure 4 and Figure 6).
The relative change in the rate of nucleation depends on the value of , which in turn, depends on the dynamic heat capacity of the glass-forming liquid. Let us denote by and the temperature perturbations with and without the influence of the dynamic heat capacity, respectively. In fact, is approximately 20–30% greater than (see Figure 1 and Figure 6). As a result, is an order of magnitude larger than . For example, in polybutylene succinate, 354 at 60 K (see Table 4) and is about 0.25 K at of about 0.25–0.3 ns (see Figure 4 and Figure 5). Therefore, the ratio is about 20. Thus, the change in the nucleation rate is significantly enhanced due to the influence of the dynamic heat capacity of glass-forming liquids.
5. Conclusions
The development of new advanced materials with artificial nanostructures requires a deep understanding of the thermal processes during the nucleation of crystals in glass-forming substances and polymers. We found that local temperature changes associated with the energy required for the formation of crystal nuclei in glass-forming liquids can be of the order of 1 K, which can significantly change the evolution of near-critical nuclei and the dynamics of nucleation. In fact, a significant number of subcritical nuclei can become stable, since the critical radius and the energy barrier decrease with decreasing local temperature, due to local energy costs for the formation of a nucleus. Local temperature perturbations during crystal nucleation in silicate glasses and polymers have been found to change the nucleation rate by 2–5 orders of magnitude. Moreover, the change in the nucleation rate is enhanced by an order of magnitude, due to the effect of the dynamic heat capacity of glass-forming liquids. The effect of the dynamic heat capacity on local thermal perturbations in glass-forming liquids can be described by an integro-differential heat equation with “memory”. This equation can be solved analytically for a spherically symmetric problem. It has been established that even fast (about 1 ns) components of the dynamic heat capacity significantly enhance local temperature perturbations arising during nucleation in glass-forming liquids. This effect is enhanced with an increase in the relaxation time of the dynamic heat capacity . The influence of saturates as increases above tens of ns. This study can be helpful for understanding the processes of nanocrystallization in glass-forming liquids and for the technology of nanostructured glasses, polymers, and composites.
Author Contributions
Conceptualization, A.M.; formal analysis, A.M.; methodology, A.M.; supervision, C.S.; visualization, A.M.; writing—original draft, A.M.; writing—review and editing, C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
A.M. acknowledges the administrative and technical support of the Prokhorov General Physics Institute of the Russian Academy of Sciences.
Conflicts of Interest
The authors declare no conflict of interest.
Nomenclature
| Latin Symbols | |
| parameter of the VFTH equation (dimensionless) | |
| and | parameters of the VFTH equation (K) |
| , | initial and equilibrium heat capacities (J·kg−1·K−1) |
| dynamic heat capacity (J·kg−1·K−1) | |
| equilibrium thermal diffusivity (m2·s−1) | |
| activation energy for the transport of segments in polymers (kJ·mol−1) | |
| kinetic barrier to nucleation (J) | |
| energy barrier for homogeneous nucleation (J) | |
| change in volumetric Gibbs free energy at crystallization (J·m−3) | |
| volumetric enthalpy of melting (J·m−3) | |
| distribution function (s−1) | |
| nucleation rate (m−3·s−1) | |
| Boltzmann constant (J·K−1) | |
| critical lateral thickness (nm) | |
| universal gas constant (J·mol−1·K−1) | |
| minimum work needed to form the nucleus (J) | |
| critical radius (nm) | |
| local change in temperature (K) | |
| undercooling (K) | |
| glass transition temperature (K) | |
| equilibrium melting temperature (K) | |
| volume of the critical nucleus (nm3) | |
| sound speed (m·s−1) | |
| Greek Symbols | |
| relaxation parameter (dimensionless) | |
| (dimensionless) | |
| viscosity (Pa·s) | |
| thermal conductivity (W·m−1·K−1) | |
| relaxation parameter (dimensionless) | |
| density (kg·m−3) | |
| σ | interfacial free energy (J·m−2) |
| interfacial free energy for fold surface (J·m−2) | |
| Debye relaxation time (s) | |
| Kohlrausch relaxation time (s) | |
| relaxation time (s) | |
| time of nucleation and decay of a subcritical nucleus (s) | |
| volumetric heat flux density (W·m−3) | |
| volumetric heat flux density (W·m−3) | |
| nth Fourier component (W·m−2) | |
| nth Fourier component (K·m) | |
| temperature modulation frequency (rad·s−1) | |
References
- Liu, H.; Jiang, Q.; Huo, J.; Zhang, Y.; Yang, W.; Li, X. Crystallization in additive manufacturing of metallic glasses: A review. Addit. Manuf. 2020, 36, 101568. [Google Scholar] [CrossRef]
- Li, X.P. Additive manufacturing of advanced multi-component alloys: Bulk metallic glasses and high entropy alloys. Adv. Eng. Mater. 2018, 20, 1700874. [Google Scholar] [CrossRef]
- Liu, H.; Yang, D.; Jiang, Q.; Jiang, Y.; Yang, W.; Liu, L.; Zhang, L.C. Additive manufacturing of metallic glasses and high-entropy alloys: Significance, unsettled issues, and future directions. J. Mat. Sci. Technol. 2022, 140, 79–120. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, H.; Mo, J.; Wang, M.; Chen, Z.; He, Y.; Yang, W.; Tang, C. Atomic-level crystallization in selective laser melting fabricated Zr-based metallic glasses. Phys. Chem. Chem. Phys. 2019, 21, 12406–12413. [Google Scholar] [CrossRef]
- Perepezko, J.H.; Gao, M.; Wang, J.Q. Nanoglass and nanocrystallization reactions in metallic glasses. Front. Mater. 2021, 8, 663862. [Google Scholar] [CrossRef]
- Zhao, B.; Yang, B.; Abyzov, A.S.; Schmelzer, J.W.P.; Rodríguez-Viejo, J.; Zhai, Q.; Schick, C.; Gao, Y. Beating homogeneous nucleation and tuning atomic ordering in glass-forming metals by nanocalorimetry. Nano Lett. 2017, 17, 7751–7760. [Google Scholar] [CrossRef]
- Wang, L.; Ma, L.; Kimura, H.; Inoue, A. Amorphous Forming Ability and Mechanical Properties of Rapidly Solidified Al–Zr–LTM (LTM=Fe, Co, Ni and Cu) Alloys. Mater. Lett. 2002, 52, 47–52. Available online: https://www.sciencedirect.com/journal/materials-letters/vol/52/issue/1 (accessed on 23 November 2022). [CrossRef]
- Mansouri, M.; Simchi, A.; Varahram, N.; Park, E.S. Development of fcc-Al nanoparticles during crystallization of amorphous Al–Ni alloys containing mischmetal: Microstructure and hardness evaluation. Mater. Sci. Eng. A 2014, 604, 92–97. [Google Scholar] [CrossRef]
- Singh, D.; Singh, D.; Mandal, R.K.; Srivastava, O.N.; Tiwari, R.S. Crystallization behavior and mechanical properties of (Al90Fe5Ce5)100−xTix amorphous alloys. J. Alloys Comp. 2016, 687, 990–998. [Google Scholar] [CrossRef]
- Lee, B.S.; Burr, G.W.; Shelby, R.M.; Raoux, S.; Rettner, C.T.; Bogle, S.N.; Darmawikarta, K.; Bishop, S.G.; Abelson, J.R. Observation of the role of subcritical nuclei in crystallization of a glassy solid. Science 2009, 326, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Orava, J.; Greer, A.L. Fast crystal growth in glass-forming liquids. J. Non Crystal. Sol. 2016, 451, 94–100. [Google Scholar] [CrossRef]
- Fokin, V.M.; Zanotto, E.D.; Yuritsyn, N.S.; Schmelzer, J.W.P. Homogeneous crystal nucleation in silicate glasses: A 40 years perspective. J. Non-Crystal. Sol. 2006, 352, 2681–2714. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Davis, G.T.; Lauritzen, J.I., Jr. The rate of crystallization of linear polymers with chain folding. In Treatise on Solid State Chemistry; Hannay, N.B., Ed.; Springer: Boston, MA, USA, 1976. [Google Scholar] [CrossRef]
- Andrianov, R.A.; Androsch, R.; Zhang, R.; Mukhametzyanov, T.A.; Abyzov, A.S.; Schmelzer, J.W.P.; Schick, C. Growth and dissolution of crystal nuclei in poly(l-lactic acid) (PLLA) in Tammann’s development method. Polymer 2020, 196, 122453. [Google Scholar] [CrossRef]
- Schick, C.; Androsch, R.; Schmelzer, J.W.P. Homogeneous crystal nucleation in polymers. J. Phys. Condens. Matter. 2017, 29, 453002. [Google Scholar] [CrossRef]
- Reiter, G.; Strobl, G.R. Progress in Understanding of Polymer Crystallization, Lecture Notes in Physics; Springer: Berlin/Heidelberg, Germany, 2007; ISBN 10: 9783540473053. [Google Scholar] [CrossRef]
- Russo, J.; Tanaka, H. The microscopic pathway to crystallization in supercooled liquids. Sci. Rep. 2012, 2, 505. [Google Scholar] [CrossRef]
- Wyslouzil, B.E.; Seinfeld, J.H. Nonisothermal homogeneous nucleation. J. Chem. Phys. 1992, 97, 2661–2670. [Google Scholar] [CrossRef]
- Birge, N.O.; Nagel, S.R. Specific heat spectroscopy of the glass transition. Phys. Rev. Lett. 1985, 54, 2674–2677. [Google Scholar] [CrossRef] [PubMed]
- Korus, J.; Beiner, M.; Busse, K.; Kahle, S.; Unger, R.D. Heat capacity spectroscopy at the glass transition in polymers. Thermochim. Acta 1997, 304–305, 99–110. [Google Scholar] [CrossRef]
- Chua, Y.Z.; Schulz, G.; Shoifet, E.; Huth, H.; Zorn, R.; Scmelzer, J.W.P.; Schick, C. Glass transition cooperativity from broad band heat capacity spectroscopy. Colloid Polym. Sci. 2014, 292, 1893–1904. [Google Scholar] [CrossRef]
- Minakov, A.; Schick, C. Nanometer scale thermal response of polymers to fast thermal perturbations. J. Chem. Phys. 2018, 149, 074503. [Google Scholar] [CrossRef]
- Landau, L.D.; Lifshitz, E.M. Course of Theoretical Physics 5: Statistical, 3rd ed.; Butterworth-Heinemann: Oxford, UK, 1980; ISBN 13: 978-0750633727. [Google Scholar]
- Landau, L.D.; Lifshitz, E.M. Course of Theoretical Physics 10: Physical Kinetics, 1st ed.; Pergamon Press: Oxford, UK; Frankfurt, Germany, 1981; ISBN 978-0-08-026480-6. [Google Scholar]
- Schmelzer, J.W.P. Application of the Nucleation Theorem to Crystallization of Liquids: Some General Theoretical Results. Entropy 2019, 21, 1147. [Google Scholar] [CrossRef]
- Kelton, K.F.; Greer, A.L. Nucleation in Condensed Matter—Applications in Materials and Biology. Pergamon Mater. Ser. 2010, 15, 1–15. [Google Scholar] [CrossRef]
- Kashchiev, D.; van Rosmalen, G.M. Review: Nucleation in solutions revisited. Cryst. Res. Technol. 2003, 38, 555–574. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 90th ed.; CRC Press: Boca Raton, FL, USA, 2010; ISBN 13: 978-1420090840/10:1420090844. [Google Scholar]
- Minakov, A.A.; Schick, C. Integro-differential equation for the non-equilibrium thermal response of glass-forming materials: Analytical solutions. Symmetry 2021, 13, 256. [Google Scholar] [CrossRef]
- Blow, K.E.; Quigley, D.; Sosso, G.C. The seven deadly sins: When computing crystal nucleation rates, the devil is in the details. J. Chem. Phys. 2021, 155, 040901. [Google Scholar] [CrossRef] [PubMed]
- Separdar, L.; Rino, J.P.; Zanotto, E.D. Unveiling nucleation dynamics by seeded and spontaneous crystallization in supercooled liquids. Comput. Mater. Sci. 2021, 199, 110802. [Google Scholar] [CrossRef]
- Tipeev, A.O.; Zanotto, E.D.; Rino, J.P. Crystal nucleation kinetics in supercooled germanium: MD simulations versus experimental data. J. Phys. Chem. B 2020, 124, 7979–7988. [Google Scholar] [CrossRef] [PubMed]
- Mahata, A.; Zaeem, M.A.; Baskes, M.I. Understanding homogeneous nucleation in solidification of aluminum by molecular dynamics simulations. Model. Simul. Mater. Sci. Eng. 2018, 26, 025007. [Google Scholar] [CrossRef]
- Fitzner, M.; Sosso, G.C.; Coxa, S.J. Michaelidesa, Ice is born in low-mobility regions of supercooled liquid water. Proc. Natl. Acad. Sci. USA 2019, 116, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Sosso, G.C.; Miceli, G.; Caravati, S.; Giberti, F.; Behler, J.; Bernasconi, M. Fast Crystallization of the Phase Change Compound GeTe by Large-Scale Molecular Dynamics Simulations. J. Phys. Chem. Lett. 2013, 4, 4241–4246. [Google Scholar] [CrossRef]
- Sosso, G.C.; Chen, J.; Cox, S.J.; Fitzner, M.; Pedevilla, P.; Zen, A.; Michaelides, A. Crystal Nucleation in Liquids: Open Questions and Future Challenges in Molecular Dynamics Simulations. Chem. Rev. 2016, 116, 7078–7116. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.M.; Baik, H.K.; Seong, B.S.; Munetoh, S.; Motooka, T. Molecular-dynamics analysis of the nucleation and crystallization process of Si. Phys. B Condens. Matter 2007, 392, 266–271. [Google Scholar] [CrossRef]
- Sanz, E.; Vega, C.; Espinosa, J.R.; Caballero-Bernal, R.; Abascal, J.L.F.; Valeriani, C. Homogeneous Ice Nucleation at Moderate Supercooling from Molecular Simulation. J. Am. Chem. Soc. 2013, 135, 15008–15017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, X. The effect of chain interpenetration on an ordering process in the early stage of polymer crystal nucleation. Polymer 2006, 47, 5213–5219. [Google Scholar] [CrossRef]
- Yamamoto, T. Molecular dynamics simulation of polymer crystallization through chain folding. J. Chem. Phys. 1997, 107, 2653–2663. [Google Scholar] [CrossRef]
- Waheed, N.; Ko, M.J.; Rutledge, G.C. Molecular simulation of crystal growth in long alkanes. Polymer 2005, 46, 8689–8702. [Google Scholar] [CrossRef]
- Gu, J.; Wang, X.; Wu, J.; Wang, X. Molecular Dynamics Simulation of Chain Folding for Polyethylene Subjected to Vibration Excitation. Int. J. Polym. Sci. 2014, 2014, 506793. [Google Scholar] [CrossRef]
- Luo, C.; Sommer, J.U. Frozen topology: Entanglements control nucleation and crystallization in polymers. Phys. Rev. Lett. 2014, 112, 195702. [Google Scholar] [CrossRef]
- Kelton, K.F. Crystal nucleation in liquids and glasses. Solid State Phys. 1991, 45, 75–177. [Google Scholar] [CrossRef]
- Rodrigues, L.R.; Fokin, A.S.A.V.M.; Zanotto, E.D. Effect of structural relaxation on crystal nucleation in a soda-lime-silica glass. J. Am. Ceram. Soc. 2021, 104, 3212–3223. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Tropin, T.V.; Fokin, V.M.; Abyzov, A.S.; Zanotto, E.D. Effects of Glass Transition and Structural Relaxation on Crystal Nucleation: Theoretical Description and Model Analysis. Entropy 2020, 22, 1098. [Google Scholar] [CrossRef]
- Rodrigues, L.R.; Abyzov, A.S.; Fokin, V.M.; Schmelzer, J.W.P.; Zanotto, E.D. Relaxation effect on crystal nucleation in a glass unveiled by experimental, numerical, and analytical approaches. Acta Mater. 2022, 223, 117458. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Tropin, T.V. Theory of crystal nucleation of glass-forming liquids: Some new developments. Int. J. Appl. Glass Sci. 2022, 13, 171–198. [Google Scholar] [CrossRef]
- Cabral, A.A.; Fokin, V.M.; Zanotto, E.D. Nanocrystallization of fresnoite glass. II. Analysis of homogeneous nucleation kinetics. J. Non Crystal. Sol. 2004, 343, 85–90. [Google Scholar] [CrossRef]
- Zanotto, E.D.; Fokin, V.M. Recent Studies of Internal and Surface Nucleation In Silicate Glasses. Phil. Trans. R. Soc. Lond. A 2003, 361, 591–613. [Google Scholar] [CrossRef]
- Granasy, L.; Igioi, F. Comparison of experiments and modern theories of crystal nucleation. J. Chem. Phys. 1997, 107, 3634. Available online: https://aip.scitation.org/doi/10.1063/1.474721 (accessed on 23 November 2022). [CrossRef]
- Zanotto, E.D.; Weinberg, M. Trends in homogeneous crystal nucleation in oxide glasses. Phys. Chem. Glasses Eur. J. Glass Sci. Technol. Part B 1989, 30, 186–192. [Google Scholar]
- Gonzalez-Oliver, C.J.R.; James, P.F. Crystal nucleation and growth in a Na2O2CaO3SiO2 glass. J. Non Crystal. Sol. 1980, 38–39, 699–704. [Google Scholar] [CrossRef]
- van Krevelen, D.W.; Nijenhuis, K.T. Properties of Polymers, 4th ed.; Elsevier Science: Amsterdam, The Netherlands, 2009; ISBN 978-0-08-054819-7. [Google Scholar]
- Mark, J.E. Polymer Data Handbook, 2nd ed.; Oxford University Press: Oxford, UK, 2009; ISBN 13: 978-0195181012/10:0195181018. [Google Scholar]
- Mark, J.E. Physical Properties of Polymers Handbook; Springer: New York, NY, USA, 2007; ISBN 978-0-387-31235-4. [Google Scholar]
- Qiu, Z.; Yang, W. Crystallization kinetics and morphology of poly(butylene succinate)/poly(vinyl phenol) blend. Polymer 2006, 47, 6429–6437. [Google Scholar] [CrossRef]
- Deng, Y.; Thomas, N.L. Blending poly(butylene succinate) with poly(lactic acid): Ductility and phase inversion effects. Eur. Polym. J. 2015, 71, 534–546. [Google Scholar] [CrossRef]
- Schick, C.; Toda, A.; Androsch, R. The narrow thickness distribution of lamellae of poly(butylene succinate) formed at low melt supercooling. Macromolecules 2021, 54, 3366–3376. [Google Scholar] [CrossRef]
- Saffian, H.A.; Yamaguchi, M.; Ariffin, H.; Abdan, K.; Kassim, N.K.; Lee, S.H.; Lee, C.H.; Shafi, A.R.; Alias, A.H. Thermal, Physical and Mechanical Properties of Poly(Butylene Succinate)/Kenaf Core Fibers Composites Reinforced with Esterified Lignin. Polymers 2021, 13, 2359. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gupta, P.K. Temperature dependence of the isostructural heat capacity of a soda lime silicate glass. J. Non Crystal. Sol. 1992, 139, 239–247. [Google Scholar] [CrossRef]
- Stebbins, J.F.; Carmichael, I.S.E.; Moret, L.K. Heat capacities and entropies of silicate liquids and glasses. Contrib. Mineral. Petrol. 1984, 86, 131–148. [Google Scholar] [CrossRef]
- Hofmeister, A.M.; Sehlke, A.; Avard, G.; Bollasina, A.J.; Robert, G.; Whittington, A.G. Transport properties of glassy and molten lavas as a function of temperature and composition. J. Volcanol. Geotherm. Res. 2016, 327, 330–348. [Google Scholar] [CrossRef]
- Kang, Y.; Lee, J.; Morita, K. Thermal Conductivity of Molten Slags: A Review of Measurement, Techniques and Discussion Based on Microstructural Analysis. ISIJ Int. 2014, 54, 2008–2016. [Google Scholar] [CrossRef]
- Sukenaga, S.; Endo, T.; Nishi, T.; Yamada, H.; Ohara, K.; Wakihara, T.; Inoue, K.; Kawanishi, S.; Ohta, H.; Shibata, H. Thermal Conductivity of Sodium Silicate Glasses and Melts: Contribution of Diffusive and Propagative Vibration Modes. Front. Mater. 2021, 8, 753746. [Google Scholar] [CrossRef]
- Berberan-Santos, M.N.; Bodunov, E.N.; Valeur, B. Mathematical functions for the analysis of luminescence decays with underlying distributions 1 Kohlrausch decay function (stretched exponential). Chem. Phys. 2005, 315, 171–182. [Google Scholar] [CrossRef]
- Ezzeddine, I.; Ghorbel, N.; Ilsouk, M.; Arous, M.; Lahcini, M.; Bouharras, F.Z.; Raihane, M.; Kallel, A. Dielectric and thermal characteristics of Beidellite nanoclay-reinforced poly (butylene succinate). Mater. Chem. Phys. 2021, 258, 123855. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).