3.2. Catalyst Characterization
The N
2 adsorption–desorption measurements were conducted to examine the textural properties of the catalysts.
Table 3 summarizes the BET specific surface area, pore volume, and pore diameter of the bare supports and synthesized samples.
The surface area decreased in the following order: NiMo/SiO
2 > NiMo/γ-Al
2O
3 > NiMo/TiO
2 which was in a similar trend corresponding with their bare supports. The surface area, total pore volume, and mean pore diameter slightly decreased after loading with NiMo for all supports. The adsorption–desorption isotherms of the prepared samples are shown in
Figure 1. It was found that the calcined NiMo/γ-Al
2O
3, NiMo/SiO
2, and NiMo/TiO
2 exhibited type IV isotherm, indicating the characteristics of a mesoporous structure. This result is consistent with the pore sizes of all the catalysts observed by the BET, which were in the range of 5–13 nm. The hysteresis loop consists of adsorption and desorption branches depending on the pore shape of the catalyst.
The phase identification and crystallinity of the synthesized catalyst were analyzed through XRD. The XRD patterns of the calcined, sulfided, and spent NiMo on γ-Al
2O
3, SiO
2, and TiO
2 catalysts are represented in
Figure 2a–c. The peaks at 2θ = 32.60°, 38.49°, 56.7°, 61.37°, and 67.94° corresponding to the (220), (222), (400), (511), and (440) phase assigned to γ-Al
2O
3 were observed for the calcined and sulfided samples as shown in
Figure 2a [
10,
11,
35]. Besides, the XRD patterns of NiMo supported on TiO
2 samples exhibited the rutile phase peak at 36° (101) and 55° (211) and anatase phase peak at 25° (101) and 48° (200), suggesting the mixed-phase of TiO
2 in this present experiment [
8,
36].
Figure 2b shows the XRD patterns of the calcined samples. It was confirmed that the MoO
2 and Mo
4O
11 phases were observed in the NiMo/SiO
2 and NiMo/TiO
2 samples [
37]. The XRD pattern of NiO [
38,
39] was found in NiMo/SiO
2. However, the MoO
3 peak could not be clearly detected for all the catalysts [
40]. After the presulfidation treatment, the characteristic peaks of MoS
2 were obviously observed over NiMo/SiO
2 (
Figure 3) at 33.5°and 58° corresponding to the (100) and (110) phase, respectively [
41,
42,
43]. On the other hand, the peaks of MoS
2 could not be observed in NiMo/γ-Al
2O
3 catalyst because this peak overlaps with the support. In the case of the NiMo/TiO
2 catalyst, the XRD pattern showed highly dispersed MoS
2 on TiO
2 support.
The H
2-TPR experiments were employed to investigate the reducibility and metal-support interaction of the prepared samples. The H
2-TPR profiles of the samples are shown in
Figure 3. The main reduction peak was located between 300 and 450 °C which might correspond to the simultaneous reduction of NiO and NiMoO
4 [
44,
45]. The TiO
2-supported catalyst exhibited a weak metal–support interaction which was the most facile to reduce (reduction temperature of 350 °C). Meanwhile, the reduction peaks of γ-Al
2O
3 and SiO
2-supported catalysts were shifted to higher reduction temperature probably due to the highly dispersed smaller-size metals on the γ-Al
2O
3 and SiO
2 support [
46], leading to the strong interaction between metal and support compared to the TiO
2. The appearance of shoulder reduction peaks between 500 and 600 °C was attributed to the several reduction steps of MoO
3, which could be involved in the reduction of MoO
3 to MoO
2 and finally MoO
2 to Mo [
44,
45,
47]. The high reduction temperatures above 650 °C were assigned to the reduction of all highly dispersed Mo species. As a result, NiMo/γ-Al
2O
3 was the most difficult one to reduce which could reach full reduction at higher 800 °C, probably due to the NiAl
2O
4 and Al
2(MoO
4)
3 formation [
44].
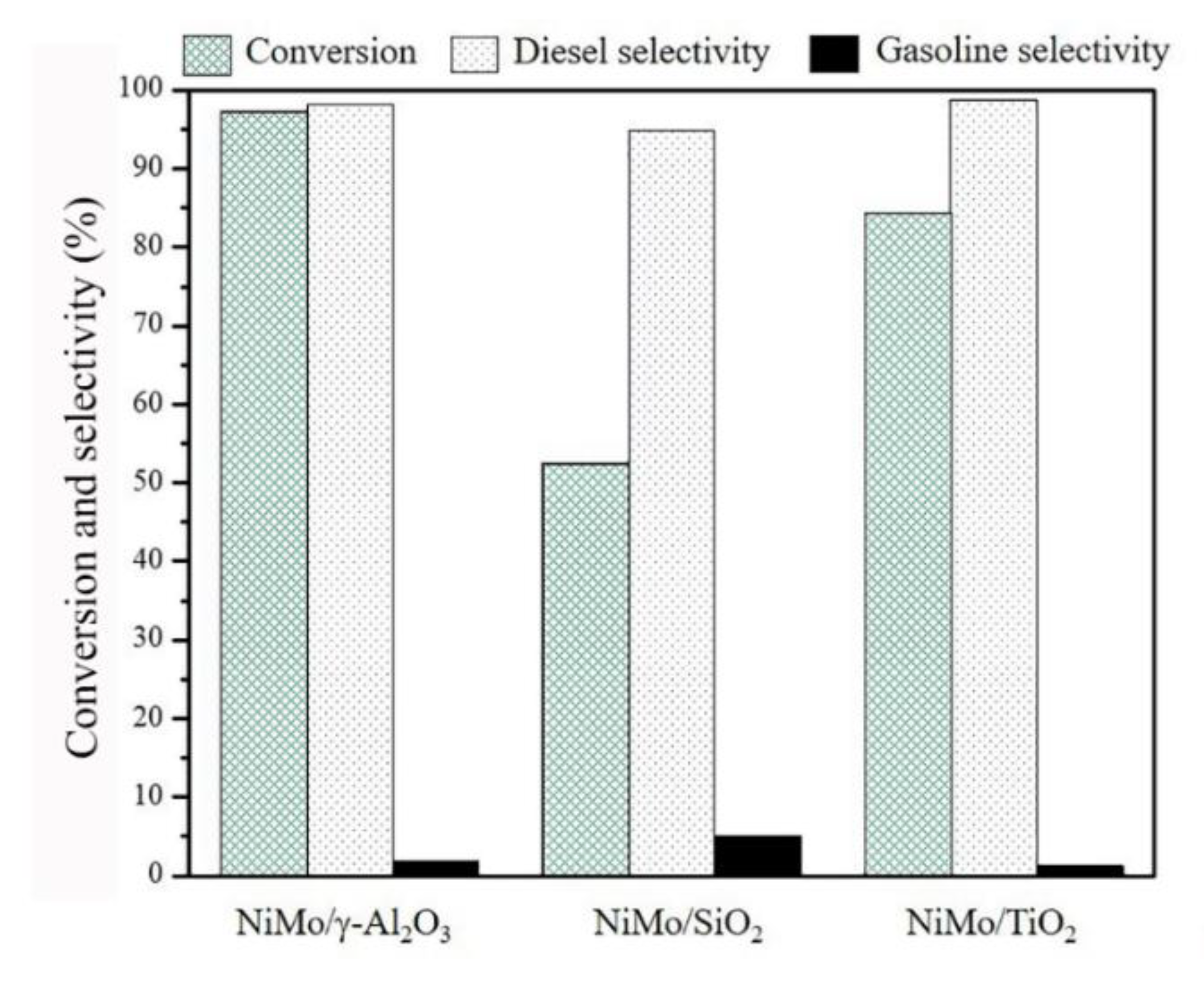
3.3. Catalytic Hydrotreating of Pongamia pinnata Oil
The triglycerides and fatty acids conversion, diesel selectivity, and gasoline selectivity over the sulfide NiMo/γ-Al
2O
3, NiMo/SiO
2, and NiMo/TiO
2 catalysts after stabilizing the reaction for 9 h are shown in
Figure 4. The conversions of
Pongamia pinnata oil of the sulfide NiMo/γ-Al
2O
3, NiMo/SiO
2, and NiMo/TiO
2 catalysts were 97.2%, 52.5%, and 84.4%, respectively. Moreover, the diesel selectivity was in a range of 95–99% for all the catalysts. The higher diesel selectivity was obtained for the sulfided NiMo/γ-Al
2O
3 and NiMo/TiO
2 catalysts compared with the sulfided NiMo/SiO
2 catalyst. Besides, the gasoline selectivity was slightly higher for the sulfided NiMo/SiO
2 catalyst (approximately 5%), suggesting the cracking reaction to lighter hydrocarbons (mixture of
n-C
5 to
n-C
11) occurred over the catalyst.
Generally, the catalytic hydrotreating process could convert the triglycerides and fatty acids into a mixture of straight-chain alkanes. The oxygen atoms in the triglycerides and fatty acids were eliminated in the forms of CO, CO2, and H2O via decarbonylation, decarboxylation, and hydrodeoxygenation, respectively. The loss of carbon atoms by CO and CO2 generation during deoxygenation can cause more mass loss of the yield of the liquid product than that of O removal via hydrodeoxygenation.
As seen in
Figure 5, the maximum theoretical yields of deoxygenated products were calculated in order to investigate the catalyst activity by assuming no side reaction occurrs. The values would be different depending on the type of starting materials and the reaction pathways (DCO/DCO
2 and HDO). In this study, the maximum theoretical yields were calculated based on the compositions of 94.3 wt.% of triglyceride and 5.7 wt.% of fatty acid contained in the extracted
Pongamia pinnata oil, which were 80.5 wt.% for DCO/DCO
2 and 85.2 wt.% for HDO. The desired product yield (diesel + gasoline) of the sulfided NiMo/γ-Al
2O
3 catalyst almost reached the theoretical value comparable to the other two catalysts, due to its high deoxygenating performance and low cracking activity.
The hydrocarbon distribution in the liquid product of sulfided NiMo/γ-Al
2O
3, NiMo/SiO
2, and NiMo/TiO
2 catalysts is summarized in
Table 4.
n-C
17 and
n-C
18 hydrocarbons are predominant hydrocarbon compositions corresponding to oleic acid, the main feed composition. Higher
n-C
18 (>50 wt.%) was observed over the γ-Al
2O
3 and TiO
2 support catalysts, suggesting that HDO reaction is preferred for these two catalysts. On the other hand, DCO and DCO
2 pathways were enhanced over the sulfided NiMo/SiO
2 catalyst as evidenced by the high amount of
n-C
17 (>60 wt.%) in the liquid product. This result can also explain the highest gas yield produced over the SiO
2-support catalyst due to the elimination of oxygen atoms in triglycerides and fatty acids in the forms of CO and CO
2.
Considering the hydrocarbon distribution above, the DCO/DCO
2 and HDO reactions occurred simultaneously over these three catalysts but with different selectivities. As obviously seen in
Figure 6, the ratio of
n-C
n-1/
n-C
n in the liquid product was examined to represent the selectivity of DCO/DCO
2 and HDO. The value of
n-C
n-1/
n-C
n decreased in the following order: NiMo/SiO
2 > NiMo/γ-Al
2O
3 > NiMo/TiO
2. For the γ-Al
2O
3- and TiO
2-supported catalysts, low ratio values (less than 0.5) were achieved, while the highest value of almost 6 was obtained over the SiO
2-supported catalyst. This can be described by the influence of Ni and Mo contents. Typically, Ni catalyst is favorable for DCO/DCO
2 pathway and also enhances the cracking reaction, higher Ni offers higher DCO/DCO
2 activity, which leads to the formation of C
n-1 hydrocarbon corresponding to the carbon contained in the feedstock. Meanwhile, the activity of the HDO reaction would be enhanced by introducing Mo species [
44,
48,
49]. Imai et al. [
49] investigated the effect of Ni, Mo, and NiMo supported on alumina for the hydrotreating of methyl laurate. They found that the product distributions were different over Ni, Mo, and NiMo catalysts. However, a sulfided NiMo/γ-Al
2O
3 catalyst exhibited the highest catalytic activity due to the synergistic effects between Ni and Mo species. In addition, the NiMo/γ-Al
2O
3 with high Mo content could enhance the conversion and the HDO reaction, as well as suppress the hydrocracking and methanation reaction.
In our investigation, the reaction was carried out with the same amount of Ni (2.4 wt.%) and Mo (9.4 wt.%) loadings; however, the product distributions were rather different. This phenomenon is probably caused by the various interactions between the supports and the metal-oxide/sulfide species. As previously stated, HDO selectivity is preferable over the MoS
2 active site; however, as seen from the TPR (
Figure 3), there was the formation of different types of metal oxide phase which could affect the MoS
2 slab formation during the sulfidation treatment, resulting in distinctions in HDO and DCO/DCO
2 selectivities, which have been extensively studied [
50,
51,
52,
53]. According to the literature, the most active phase, Type II, which is highly stacked and totally sulfided, can be formed at a weak metal–support interaction, while Type I, which is less stacked and partially sulfided, can be formed by strong interaction with the support. Based on our results, the TiO
2-supported catalyst exhibited less metal–support interaction, resulting in more MoS
2 active phase formation which leads to relatively high HDO activity compared to the other two catalysts. Meanwhile, the DCO/DCO
2 selectivity was highly active over the SiO
2-supported catalyst which could be ascribed to the MoS
2 suppression by the strong metal–support interaction.
Interestingly, the SiO
2-supported catalyst contains a relatively high surface area (see
Table 1) which could be beneficial to catalytic activity by increasing the exposure of the active sites [
46]. However, the conversion of triglyceride and fatty acid over the SiO
2-supported catalyst is relatively low compared to the value obtained from the γ-Al
2O
3 and TiO
2-supported catalysts. This result may be caused by the specific properties of the supporting materials. The deoxygenation activity also involves an acidity property of the catalyst. Mild acidity is required for the high deoxygenation activity, due to its favorable adsorption of triglycerides, fatty acid, and other oxygen-containing molecules [
54,
55,
56], whereas the strong acidity promotes unwanted hydrocracking and increases coke formation, leading to catalyst deactivation [
57]. There have been several reports indicating that high acidity support has a beneficial effect on hydrodeoxygenation activity. Kumar et al. [
38] studied the impact of Ni supported on γ-Al
2O
3, SiO
2, and HSZM-5 zeolite on stearic acid deoxygenation. They observed that utilizing the HSZM-5 catalyst resulted in the maximum conversion, which was attributed to the relatively strong acidity of the HSZM-5 catalyst compared to that of γ-Al
2O
3 and SiO
2. This consequence was also observed by Peng et al. [
58]. They studied the deoxygenation of palmitic acid with Ni supported over γ-Al
2O
3, SiO
2, and ZrO
2 and determined that conversion proceeded in the following order: ZrO
2 > γ-Al
2O
3 > SiO
2 due to the greater acidity of the ZrO
2 support. According to the literature, infrared spectroscopy of adsorbed CO [
59] and NH
3-temperature-programmed desorption [
60] were used to study the acidity of SiO
2. There were no substantial numbers of acid sites for the SiO
2 support. While the three varieties of Lewis acid sites (weak, medium, and strong) were found on γ-Al
2O
3 support [
57,
59,
61], the medium-weak forms of Lewis acid were found on anatase TiO
2 support [
54,
55]
. Taking into account all of these remarks, the XRD results revealed that the TiO
2 employed in this work had a mixed-phase (anatase + rutile); consequently, the acidity of the mixed-phase TiO
2 was apparently lower than that stated in the literature. It is worth noting that bare Al
2O
3 and TiO
2 have been reported to exhibit conversion and hydrodeoxygenation activity due to their acid-based characteristics; however, their activity is much higher once loaded with active metal [
62,
63,
64,
65]. While bare SiO
2 poses very low acidity (inertness) and no significant activity has been observed when employing bare SiO
2.
As a result, it might be speculated that the acidity of the catalyst increased in the following order: NiMo/SiO2 < NiMo/TiO2 < NiMo/γ-Al2O3, which corresponds to our conversion (NiMo/SiO2 < NiMo/TiO2 < NiMo/γ-Al2O3). Consequently, we hypothesize that the observed disparities in reaction pathways are not only driven by the type of active phase but also the acidity of the supporting materials.
Figure 7 shows the gas product compositions which mainly consist of CO, CO
2, and CH
4. The CO and CO
2 could be generated via DCO and DCO
2, whereas CH
4 could be produced from the cracking of products in the liquid phase and also a methanation reaction in the gas phase. Based on the equilibrium constant for reverse water gas shift (RWGS) reaction, the calculated value at 330 °C is approximately 0.04, suggesting that the RWGS can be neglected to convert CO
2 to CO. Hence, CO would be a primary gas product which is mainly generated from the oxygen removal of triglycerides and fatty acid via DCO. Therefore, the level of CO and CO
2 content can elucidate the activity of DCO and DCO
2 reactions of the three catalysts [
8]. By comparison, the CO level was higher than the CO
2, indicating that DCO has higher activity than DCO
2 for all the catalysts. Due to the low methane percentage in the gaseous product, the methanation process might be minimal. However, a small amount of methane was generated when the NiMo/γ-Al
2O
3 catalyst was in use. This might be attributed to the high acidity of γ-Al
2O
3 support, which enhances the adsorption of triglyceride and fatty acid molecules in feedstock, potentially accelerating methane generation. This finding might support our previous assumption concerning the acidity of the catalysts. It is worth noting that the amount of methane from NiMoS of all these supports is much less pronounced than single Ni [
66] or single Mo [
67] in various active forms.
In order to confirm the elimination of oxygen atoms in triglycerides and fatty acids, the functional groups of the liquid products were characterized by Fourier transform infrared (FTIR) spectroscopy (see
Figure 8). The absorption peak at 1704 cm
−1 in the FTIR spectrum corresponds to the carboxylic functional groups of fatty acids. On the other hand, the absorption peaks at 1746 and 1164 cm
−1 in the FTIR spectrum are assigned to the carbonyl groups (–C=O stretch) and ester groups (–C–O– stretch), respectively [
41]. In the hydrotreating process, triglyceride was firstly hydrogenolysed to fatty acids and further deoxygenated to
n-alkane. The FTIR spectrum of
Pongamia pinnata oil exhibited the absorption peaks corresponding to triglyceride and fatty acids. Considering the liquid product after the hydrotreating process, the peaks intensity of triglyceride and fatty acid in the FTIR spectrum significantly decreased over sulfided NiMo supported on γ-Al
2O
3 and TiO
2 catalysts. This result suggested that the triglyceride and fatty acid were almost completely converted to
n-alkanes. On the other hand, the peak intensity of fatty acid was detected in the liquid product when the reaction was catalyzed by the sulfided NiMo/SiO
2 catalyst.
The results of the elemental composition (CHNS/O) of extracted
Pongamia pinnata oil and liquid products are summarized in
Table 2. Thereafter the deoxygenation activity, and percent of carbon atom content in liquid product increased with decreasing oxygen concentration. The deoxygenation degree calculated based on the oxygen content of feedstock and liquid product is increased as follows: NiMo/SiO
2 (37.7%) < NiMo/γ-Al
2O
3 (94.6%) < NiMo/TiO
2 (98.3%). These results are attributed to the various intensities of metal-support interaction, as mentioned earlier. However, the relatively high catalytic performance of the NiMo/γ-Al
2O
3 compared to NiMo/TiO
2 may be due to its higher surface area and greater mesopore size.
The H/C and O/C ratios were shown on the Van Krevelen diagram (
Figure 9) to compare the elemental compositions of hydrotreated liquid products and extracted oil with petroleum fuel and hydrotreated vegetable oil (HVO) from Neste Oil.
Pongamia pinnata oil had the highest O/C ratio of 0.08, which is consistent with the high amount of oxygen-containing molecules (triglycerides and fatty acid) content. In comparison to the starting feedstock, the hydrotreated products of the three catalysts displayed greater H/C and lower O/C values, particularly for the γ-Al
2O
3 and TiO
2-supported catalysts, which were close to the value of petroleum-based oil (H/C = 1.6–1.8, O/C almost nil) [
68,
69]. Furthermore, the H/C value of NiMo/γ-Al
2O
3 (H/C = 2.17) is comparable to the value of HVO generated from Neste oil (H/C = 2.15) [
70]. The SiO
2-supported catalyst had a much higher O/C value than the other catalysts, which could be attributed to poor deoxygenation activity and strong cracking activity, as evidenced by the high gas and short hydrocarbon fraction generated. While a high H/C value ratio suggested the production of a liquid product with a high hydrogen content, this might be attributed to high hydrogenation and low cyclization/aromatization activities.
Surprisingly, slightly higher amounts of sulfur content were detected in the hydrotreated liquid products. They were 2.65%, 2.07%, and 2.56% for the NiMo/γ-Al
2O
3, NiMo/SiO
2, and NiMo/TiO
2 catalysts, respectively (
Table 2). It is even higher than the value detected in the extracted
Pongamia pinnata oil (2.16%). This result could demonstrate that there was some sulfur leaching from the NiMoS catalysts during the reaction, as the amount of sulfur content decreased after the reaction process (
Table 5). In addition, the loss of sulfur in the spent catalysts, on the other hand, could be accounted for by the oxidation of the sulfide phase, which would be incorporated by oxygen atoms at the edge sulfur atoms in the MoS
2 during the reaction.
3.4. Catalyst Stability and Deactivation
The catalytic stability of the NiMo/γ-Al
2O
3, NiMo/SiO
2, and NiMo/TiO
2 catalysts was evaluated through 15 h time-on-stream using extracted
Pongamia pinnata oil at a temperature of 330 °C, H
2 pressure of 50 bar, WHSV of 1.5 h
−1, and H
2/oil ratio of 1000 cm
3/cm
3. As can be seen in
Figure 10a, The NiMo/γ-Al
2O
3 exhibited the highest conversion and stability for 15 h, meanwhile, the NiMo/SiO
2 and NiMo/TiO
2 catalysts gradually decreased in conversion. On the other hand, unexpectedly, the NiMo/γ-Al
2O
3 and NiMo/SiO
2 catalysts showed good catalytic activity (100% conversion) and remained stable for 42 h when using refined palm olein as feedstock (
Figure 10b). This may be caused by the impurities in the
Pongamia pinnata oil (
Table 6) which were quantified using the inductively coupled plasma (ICP) technique. According to Kubicka and Horacek [
71], an abundance of alkali content in feedstock may have a deleterious influence on the catalyst. They discovered that the presence of alkalis (Ca, K, Na, and Mg) in waste rapeseed oil increased the rate of CoMoS/γ-Al
2O
3 catalyst deactivation due to their deposition on the catalyst surface, resulting in active site blockage/poisoning. Similarly, Arora et al. [
72] studied the effect of potassium-containing feedstock and discovered that when the quantity of potassium is high, the catalyst activity is inhibited. This is due to potassium atoms preferentially adsorbed on the edge vacancy sites of the MoS
2 slabs.
The effect of potassium doping on MoS
2 for CO hydrogenation has also been studied using DFT calculation by Andersen et al. [
73]. Their findings revealed that doping K on MoS
2 enhances surface basicity owing to increased electron charge, and also blocks the Mo and S edge, inhibiting CO adsorption and limiting hydrogen accessing the MoS
2 surface. In agreement with May et al. [
74,
75], they found that modifying the CoMoS/Al
2O
3 commercial catalyst with potassium resulted in decreased hydrogenation activity for a synthetic FCC gasoline, due to changing electronic properties of sulfide phase and also decreasing the acid site of support. Furthermore, calcium has been correlated to a reduction in catalyst activity, particularly HDS and HDO activities, as well as a minor decrease in hydrogenation activity [
76]. Comparing these results between using
Pongamia pinnata oil and refined palm olein, it is presumed that the suppression on catalytic activity is considerably more pronounced with NiMo/SiO
2 than with NiMo/γ-Al
2O
3 and NiMo/TiO
2. These results were probably caused by the partially facilitated by the γ-Al
2O
3 and TiO
2 and catalyzed by their acid sites. Furthermore, the prolonged activity of NiMo/γ-Al
2O
3 for
Pongamia pinnate oil was tested, which revealed that the catalyst activity dropped after 75 h, while the stability of the catalyst for refined palm olein was still longer, over 75 h.
Taking into account the presence of phosphorus (P) in feedstock, it was reported to promote oligomerization/polymerization reactions, resulting in increased formation of high molecular weight compounds, eventually leading to increased carbonaceous deposition on the surface of the catalyst and rapid catalyst deactivation [
71,
78]. As can be seen in
Figure 11, significantly greater carbonaceous deposition was discovered during the hydrotreating of
Pongamia pinnata oil than in the hydrotreating of refined palm olein in our study. TGA was performed in atmospheric air to quantify the formation of carbonaceous species on the surface of catalysts. The first weight loss at temperatures below 200 °C can be ascribed to evaporation of moisture adsorbed, whereas temperatures between 300 and 450 °C can be attributed to volatile substance decomposition. At higher temperatures ranging from 450 to 800 °C, it is ascribed to the decomposition of various carbonaceous species including heavy hydrocarbons accumulated inside the catalyst pores or more stable amorphous coke (450–500 °C), as well as the oxidation of graphite and graphene carbon (600–800 °C) [
79,
80]. The decomposition of a substrate at a temperature above 800 °C could be related to the oxidation of inorganic carbon (i.e., CaCO
3 [
80] and K
2CO
3 [
81]). Evidently, larger weight losses were detected in the catalyst employed for
Pongamia pinnata oil, especially when compared to the SiO
2- and TiO
2-supported catalysts, showing that the presence of phosphorus content might accelerate the rate of coke formation.
Ultimately, it is possible to conclude that differences in catalytic performance are caused not only by different characteristics of metals and supporting materials but also by the purity of feedstock, which appears to have a significant impact on declining catalytic activity and catalyst deactivation. Catalyst deactivation during 42 h of time-on-stream when using refined feed (refined palm olein) could be negligible for all three catalysts. However, the difference in deactivation was more pronounced when being tested under unrefined with high metal content, e.g., P. pintnata oil. Though tested under unrefined is found to be an aggressive condition for coking, it is likely that catalyst poisoning and coking is not the main cause of the deactivation.



 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}