

The Role of Carbonate Formation during CO2 Hydrogenation over MgO-Supported Catalysts: A Review on Methane and Methanol Synthesis




Abstract
:1. Introduction
2. Magnesium Oxide—Occurrence, Production and Properties
2.1. Occurrence of MgO
2.2. Production of MgO
2.3. Properties of MgO
- (i)
- Caustic MgO is formed when the solid minerals Mg(OH)2 or MgCO3 are slightly heated above their decomposition temperature. Caustic MgO can be divided into two types: light-burned and hard-burned MgO. Light-burned MgO, generally known as caustic calcined MgO, is formed at calcination temperatures of 1143–1273 K and is the most reactive form of MgO. Hard-burned MgO, which is calcined at temperatures of 1823–1923 K, has limited reactivity. The reactivity of caustic MgO decreases with increasing calcination temperature.
- (ii)
- Sintered or dead-burned MgO, also known as magnesia clinker, is produced at calcination temperatures of 1673–2273 K. It is an unreactive form of MgO with a high heat capacity and a high thermal conductivity, thus, generally being used as refractory material.
- (iii)
- Fused MgO is generally produced from naturally occurring MgCO3 in electric arc furnaces at 1273–1673 K (‘dead burned’) or by melting caustic magnesia. Fused magnesia is a crystalline substance with a melting point of 3073 K. When it is heated up to the melting point, no phase change takes place. Its tendency to undergo hydration is much lower than that of sintered or caustic calcined MgO, which makes it essentially stable toward the atmosphere. In a reducing atmosphere, the stability of fused magnesia is limited to 1973 K. Because it combines high electrical resistance and high thermal conductivity, it is mainly used as an insulating material [17].
3. CO2 Adsorption Behavior on MgO
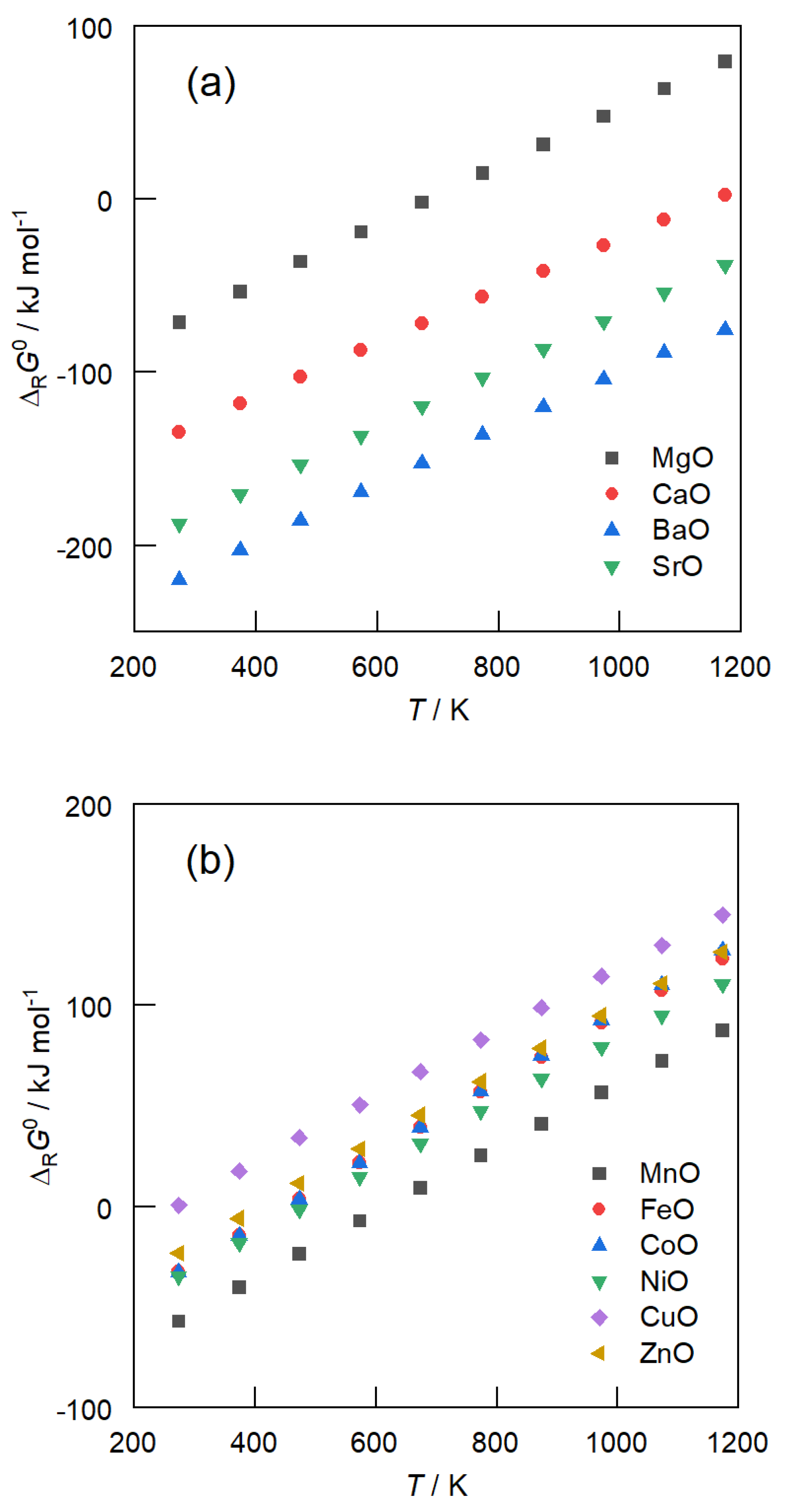
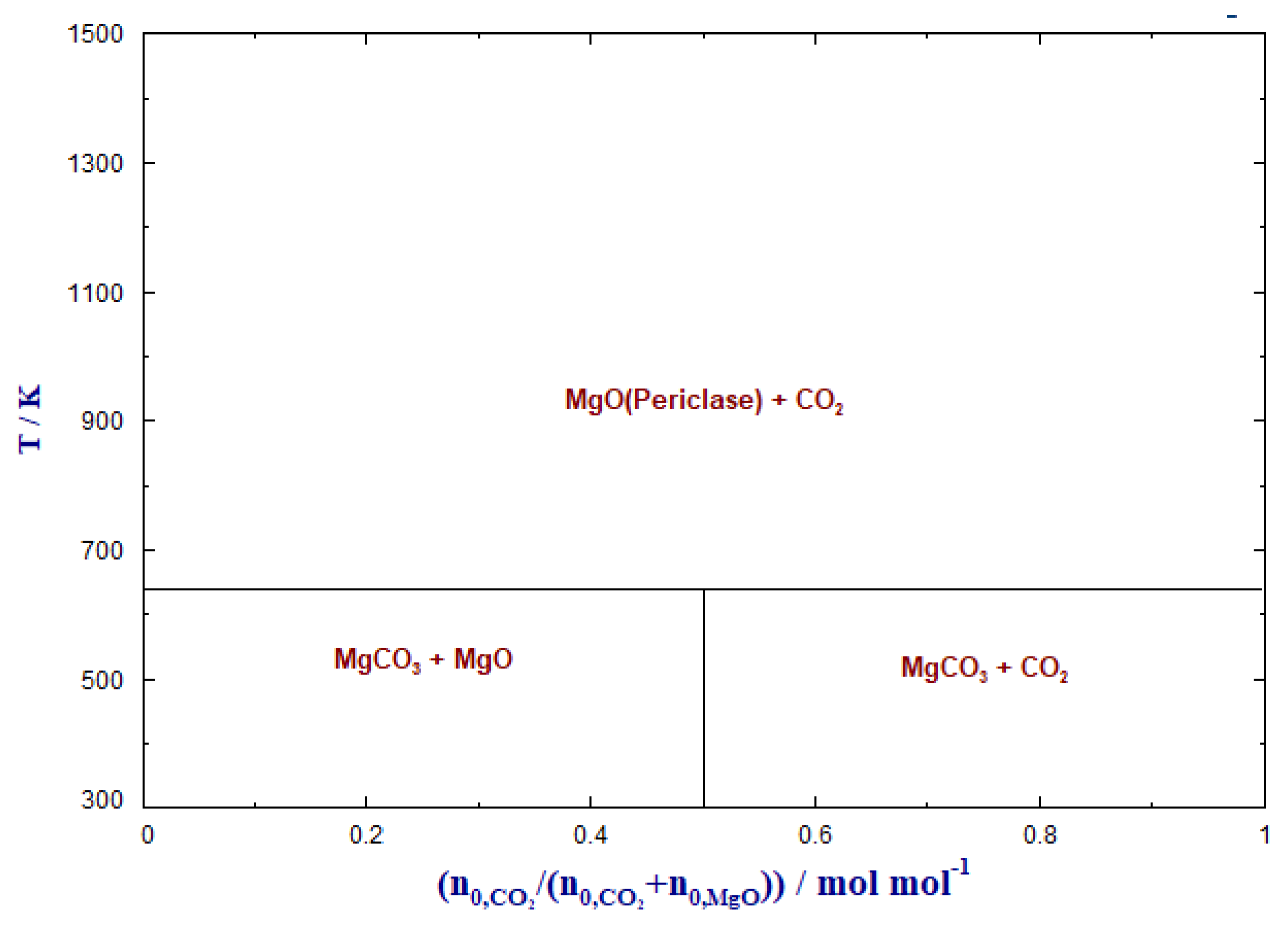
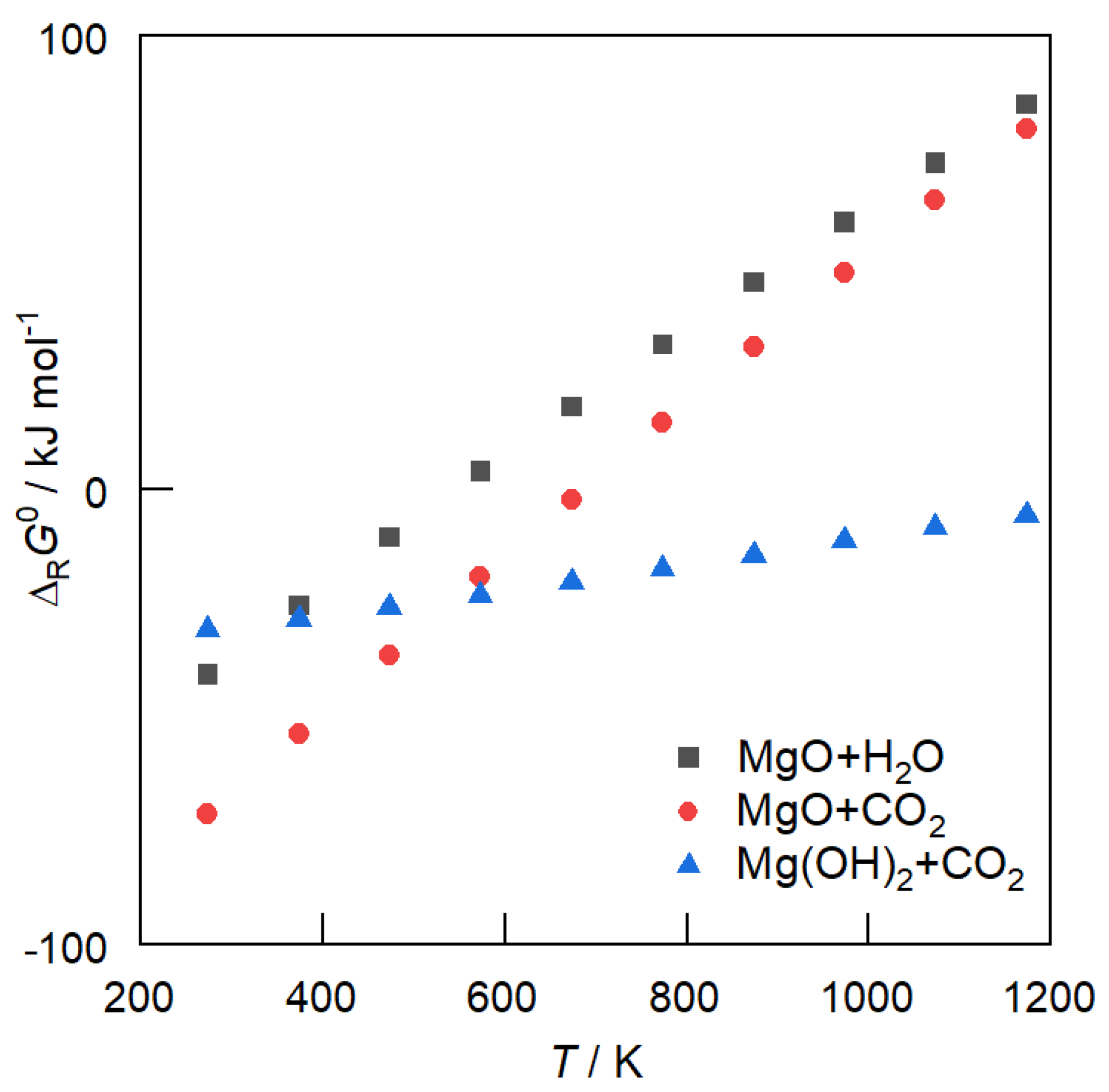
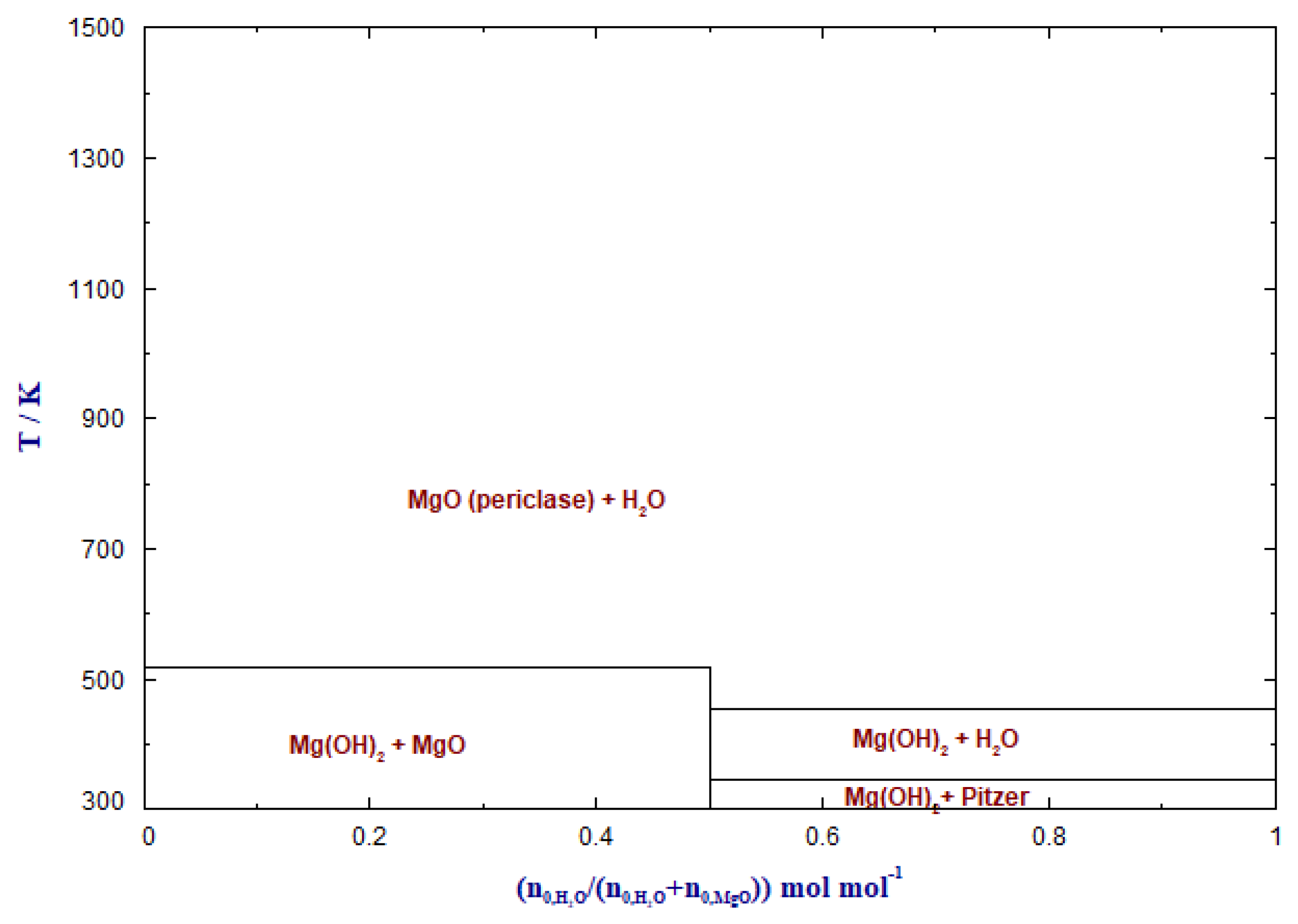
3.1. Carbonate Formation—Thermodynamic Considerations
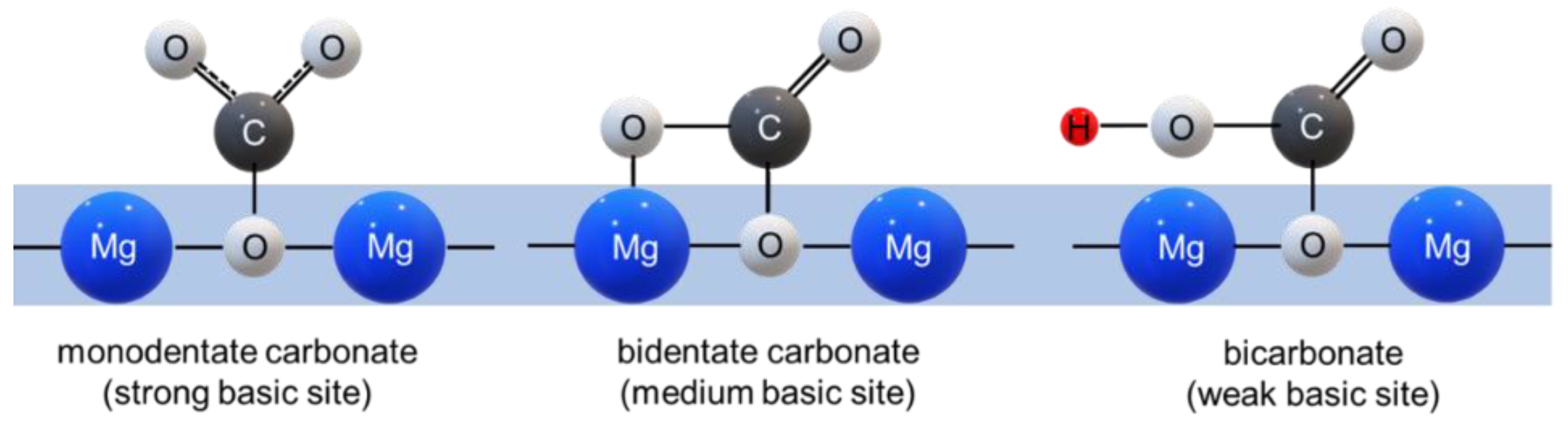
3.2. Mechanism of CO2 Adsorption on MgO
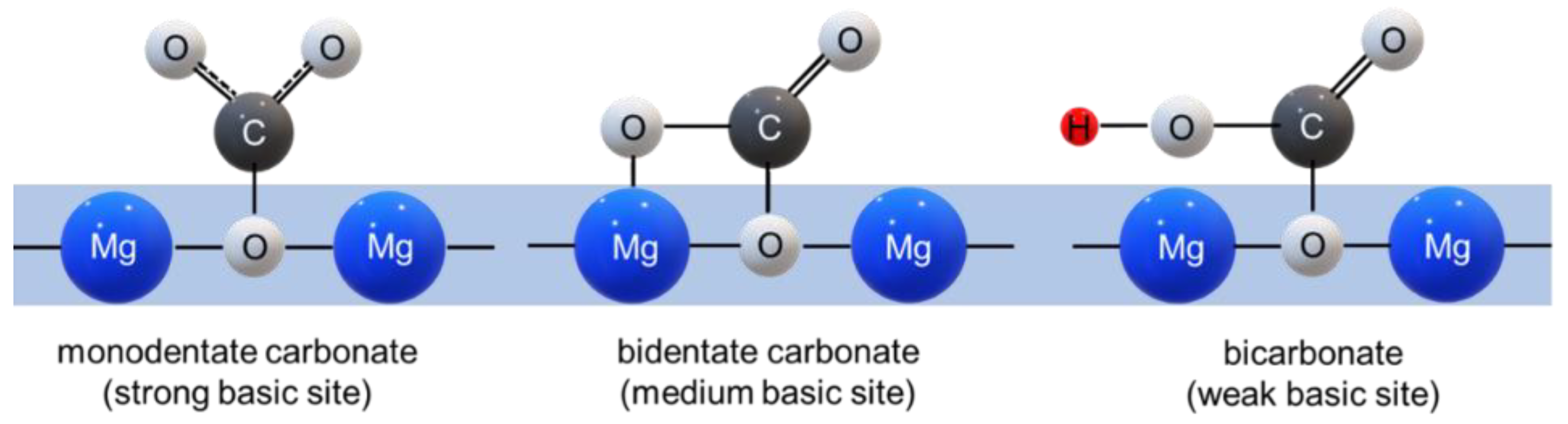
3.3. CO2 Adsorption Studies on MgO
3.3.1. Composite Promoters in MgO-Based Adsorbents
3.3.2. Effect of the Adsorption Temperature
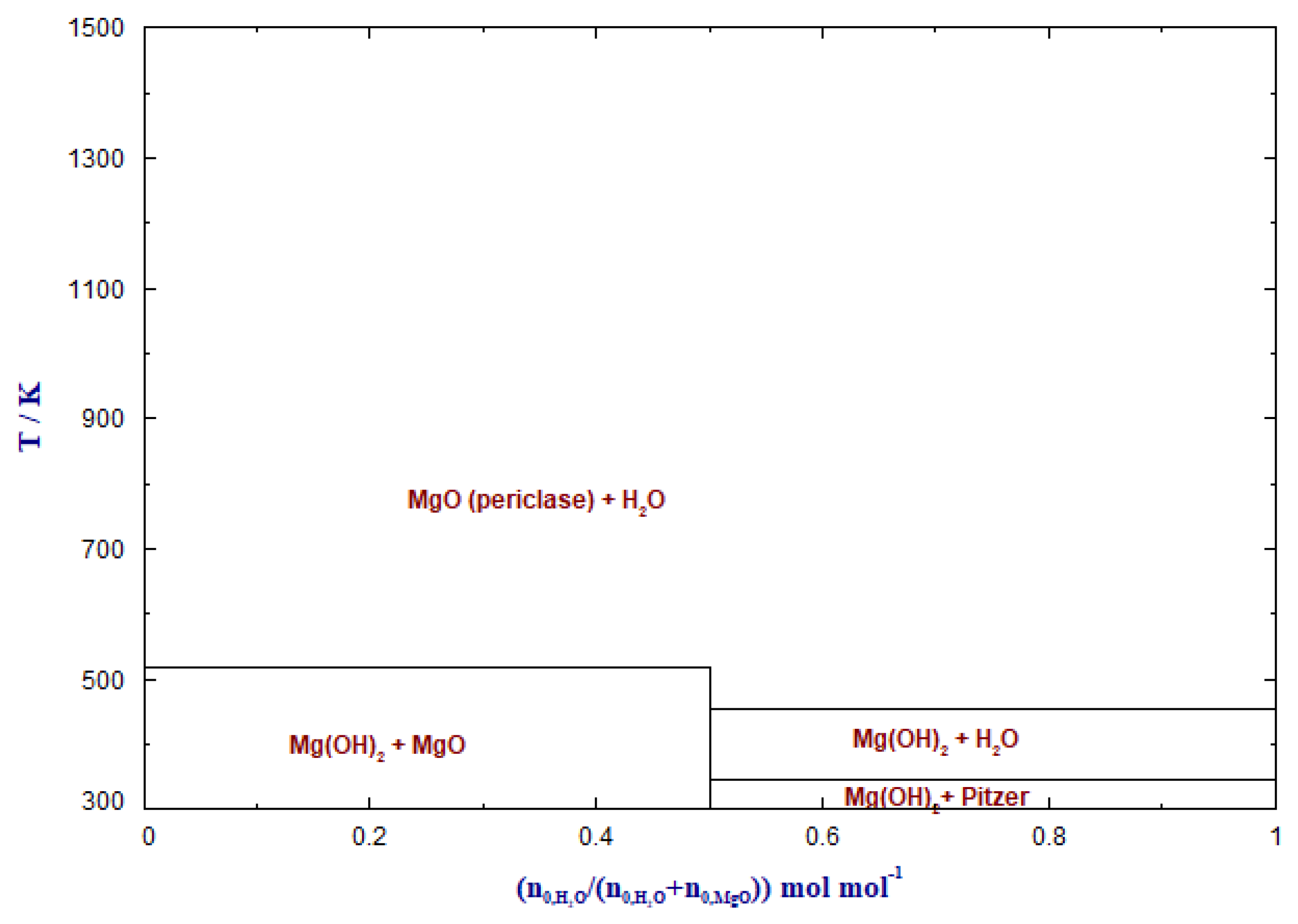
3.4. CO2 Adsorption in the Presence of Water
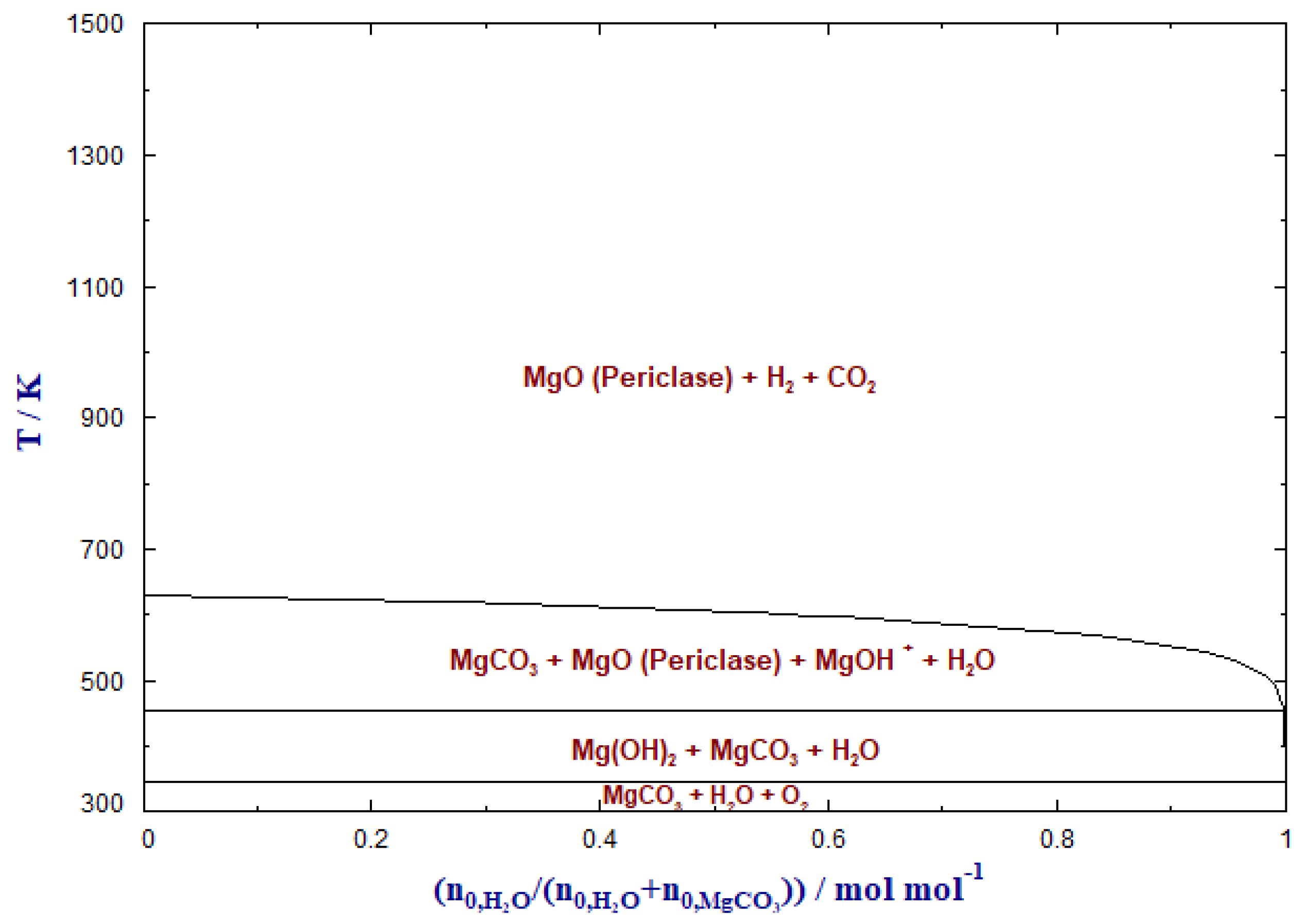
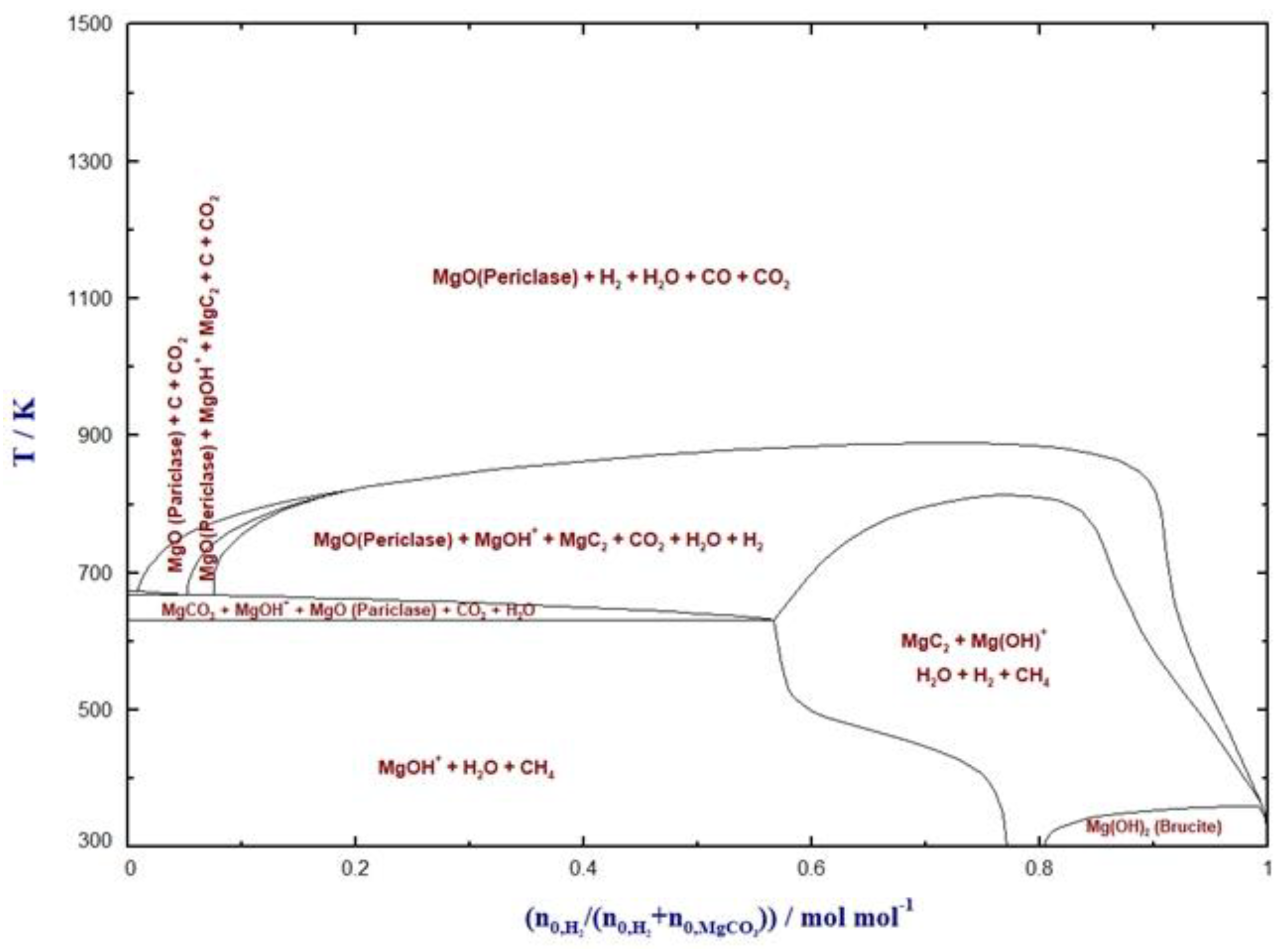
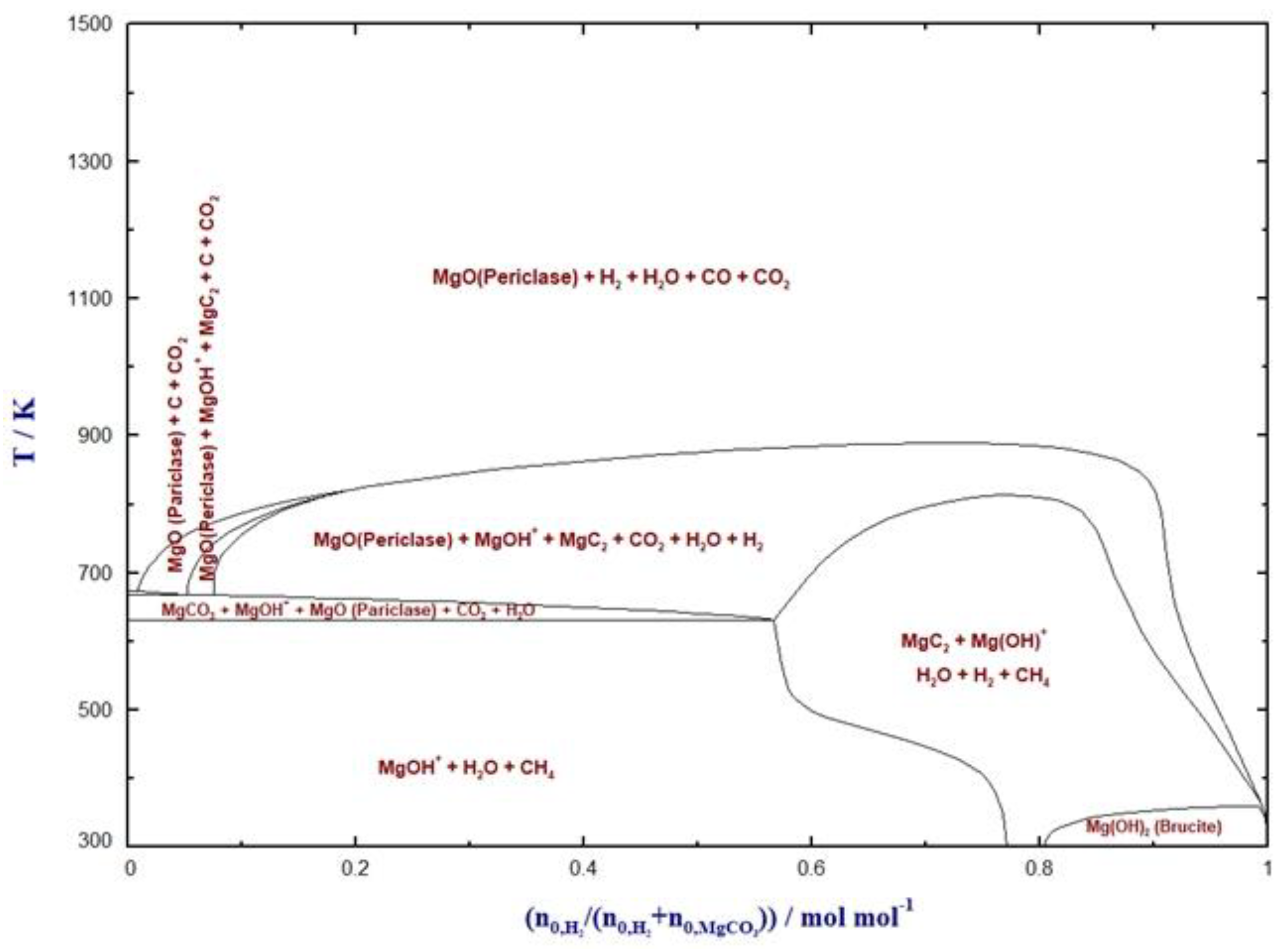
4. Hydrogenation of MgCO3
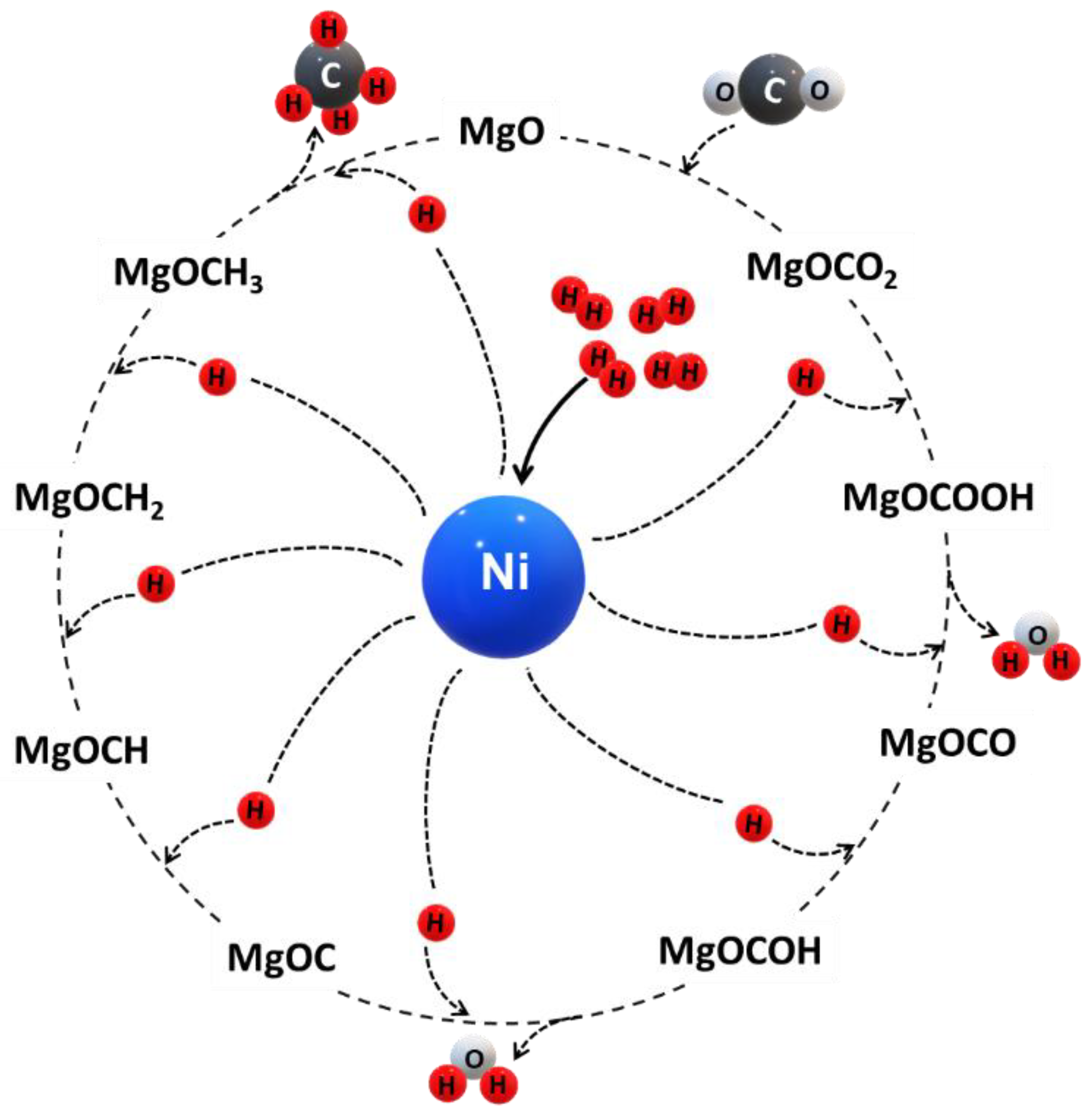
5. Methane Synthesis over MgO-Supported Catalysts
5.1. Experimental Studies for CO2 Methanation over Ni/MgO Catalysts
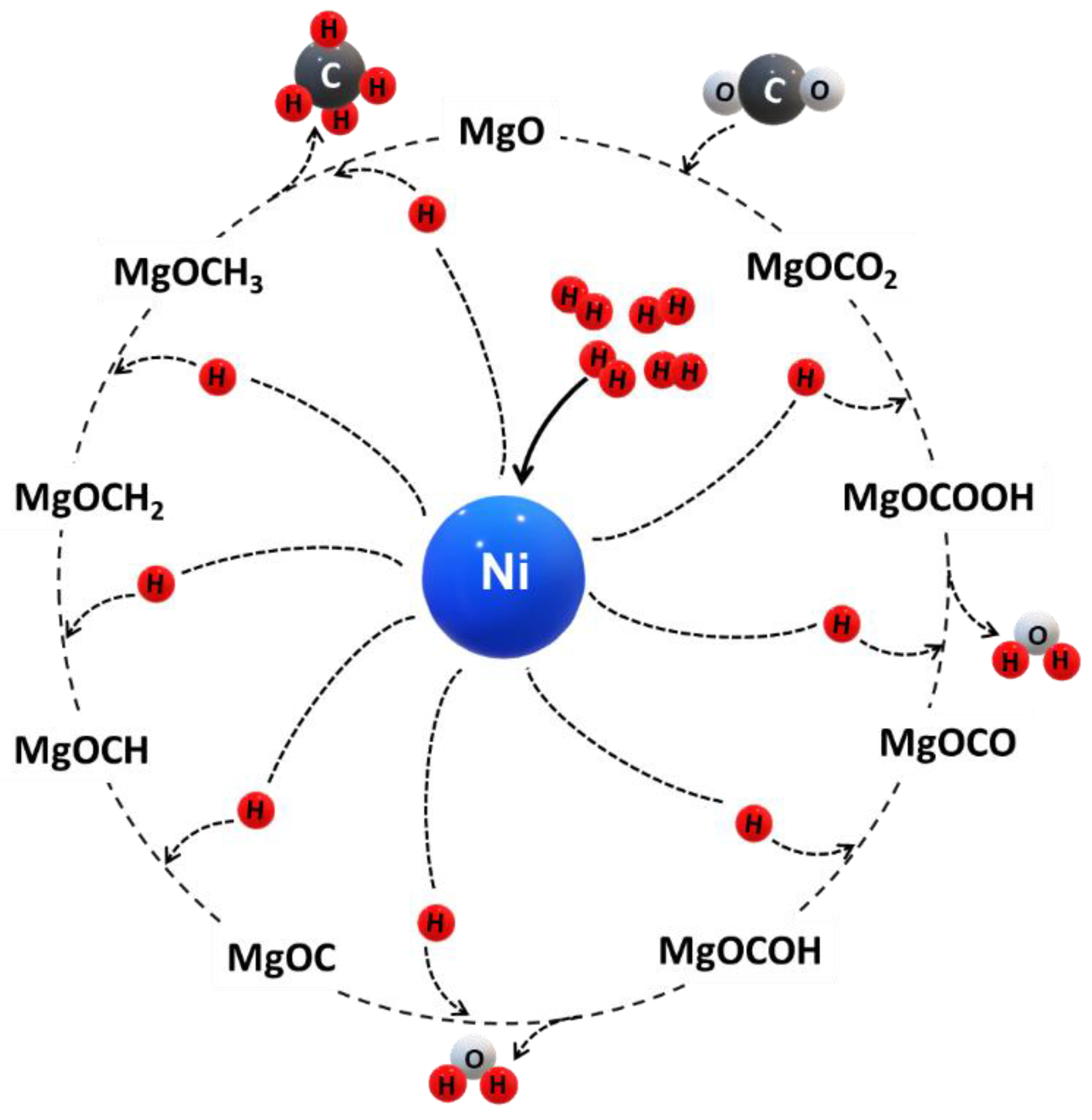
5.2. Reaction Mechanism for CO2 Methanation
- (i)
- CO2 is stabilized by MgO via CO intermediates forming carbonate at the surface. The carbonate formation has a critical role in CO2 methanation. CO dissociation may be rate-determining.
- (ii)
- The carbonate is sequentially hydrogenated producing a carboxy group and water as byproduct.
- (iii)
- The carboxy group reacts with MgO to form MgCOO and is sequentially hydrogenated forming MgCOOH, whereas one molecule of water is generated.
- (iv)
- Hydrogenation with three hydrogen atoms on the MgO surface forming MgOC—MgOCH—MgOCH2—MgOCH3, respectively.
- (v)
- MgOCH3 is hydrogenated with a hydrogen atom to form MgO and methane.
6. Methanol Synthesis over MgO-Supported Catalysts
6.1. Experimental Studies for CO2 Hydrogenation to Methanol over Cu-Based Catalysts with MgO

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Composition | Preparation Method | Operation Conditions | Performance | Comments | Ref. |
|---|---|---|---|---|---|
| CuO/MgO (wCu = 38 wt%) | impregnation | T = 573 K p = 50 bar v(CO2):v(H2) = 1:3 | XCO2 = 23–29% YCH3OH ~ 28% (after 53 h) SCH3OH ~ 88% (after 53 h) XCO2 = 76% SCH3OH = 59% |
Semi-continuous operation | [117] |
| Cu/MgO | DFT calculations | T = 500–600 K PH2 = 30 bar, PCO2 = PCO = 10 bar | - | [116] | |
| Cu/MgO (wCu = 50 wt%) | precipitation |
T
= 523 K p = 50 bar, H2/CO2/inert = 68/3/29 | YCH3OH = ~20% | [6] | |
| Cu/MgO/Al2O3 Cu:MgO:Al2O3 = 50:30:20 | co-precipitation |
T
= 523 K p = 20 bar n(H2): n(CO2) = 3:1 GHSV = 2000 h−1 and 6000 h−1 | XCO2,2000h−1 = ~3% CH3OH2000h−1 = 0.80 molCH3OH kg−1 h−1 CH3OH6000h−1 = 1.48 molCH3OH kg−1 h−1 TOFCH3OH = 11.9 × 10−4 s−1 | [113] | |
| Cu-ZnO-ZrO2-MgO/Al2O3 | impregnation |
T
= 523 K, p = 20 bar, n(H2): n(CO2) = 3:1, GHSV = 1400 h−1 | XCO2 = 12.1% SCH3OH = 36.0% SCH4 = 2.4% SCO = 61.61% STY (31.0 g kgCat−1 h−1) | mCuO/MgO = 5 g Cu:Zn:Zr:Mg = 2: 1: 0.9: 0.1 | [105] |
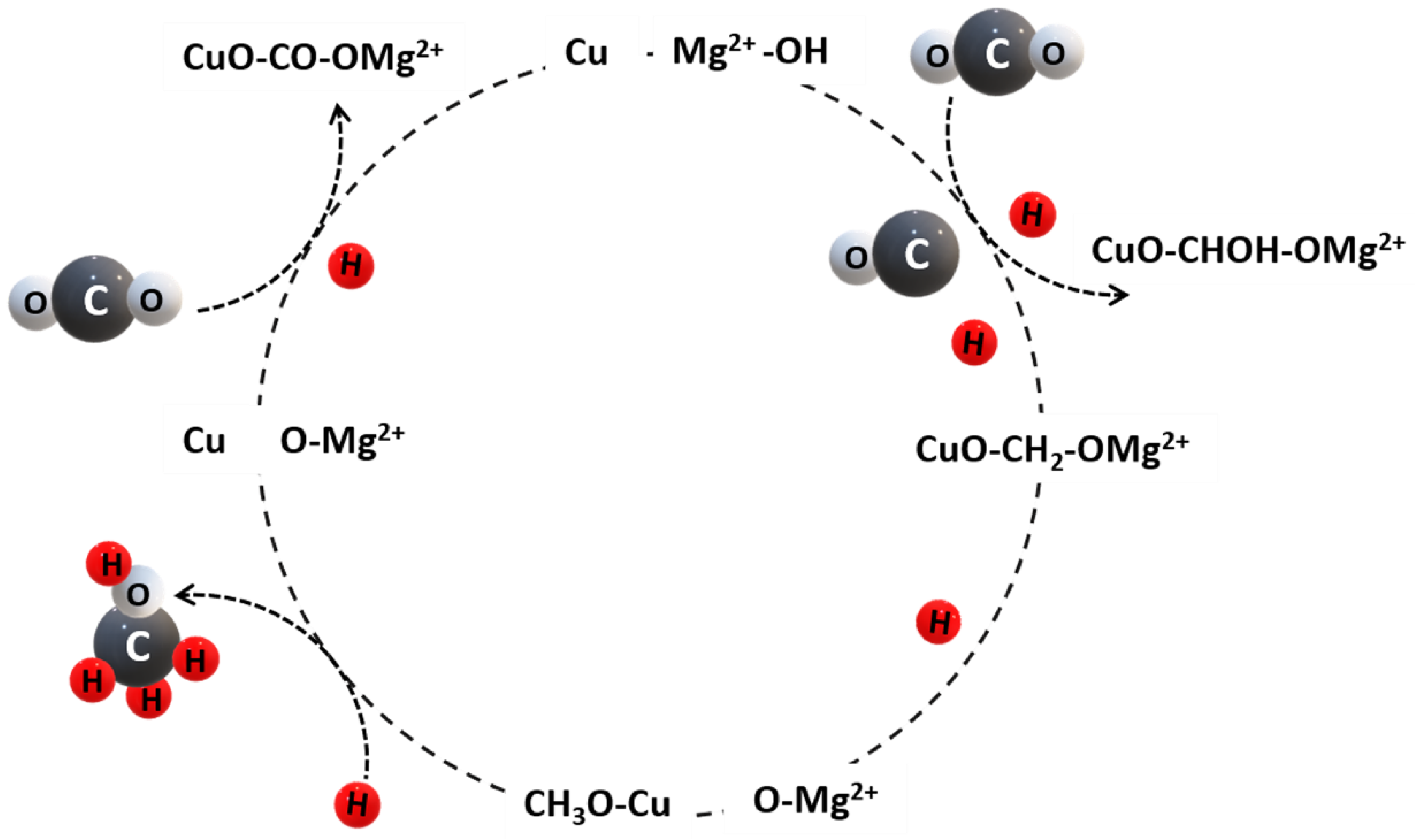
6.2. Reaction Mechanism for CO2 Hydrogenation to Methanol over Cu-Based Catalysts
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Energy Agency. Global Energy Review: CO2 Emissions in 2021. 2022. Available online: https://www.iea.org/reports/global-energy-review-co2-emissions-in-2021-2 (accessed on 21 March 2023).
- Topham, S.; Bazzanella, A.; Schiebahn, S.; Luhr, S.; Zhao, L.; Otto, A.; Stolten, D. Carbon Dioxide. Ullmann’s Encycl. Ind. Chem. 2014, 1–43. [Google Scholar] [CrossRef]
- Wei, W.; Jinlong, G. Methanation of carbon dioxide: An overview. Front. Chem. Sci. Eng. 2011, 5, 2–10. [Google Scholar] [CrossRef]
- Guil-López, R.; Mota, N.; Llorente, J.; Millán, E.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Methanol synthesis from CO2: A Review of the latest developments in heterogeneous catalysis. Materials 2019, 12, 3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent advances in carbon dioxide hydrogenation to methanol via heterogeneous catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef]
- Nielsen, N.D.; Thrane, J.; Jensen, A.D.; Christensen, J.M. Bifunctional synergy in CO hydrogenation to methanol with supported Cu. Catal. Lett. 2020, 150, 1427–1433. [Google Scholar] [CrossRef]
- Joo, O.-S.; Jung, K.-D.; Moon, I.; Rozovskii, A.Y.; Lin, G.I.; Han, S.-H.; Uhm, S.-J. Carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction (the CAMERE process). Ind. Eng. Chem. Res. 1999, 38, 1808–1812. [Google Scholar] [CrossRef]
- Goehna, H.; Koenig, P. Producing methanol from CO2. ChemTech 1994, 24. Available online: https://www.osti.gov/biblio/7157792 (accessed on 21 March 2023).
- Chatterjee, R.; Kuld, S.; van den Berg, R.; Chen, A.; Shen, W.; Christensen, J.M.; Jensen, A.D.; Sehested, J. Mapping support interactions in copper catalysts. Top. Catal. 2019, 62, 649–659. [Google Scholar] [CrossRef]
- Shen, L.; Xu, J.; Zhu, M.; Han, Y.-F. Essential Role of the Support for Nickel-Based CO2 Methanation Catalysts. ACS Catal. 2020, 10, 14581–14591. [Google Scholar] [CrossRef]
- Julkapli, N.M.; Bagheri, S. Magnesium oxide as a heterogeneous catalyst support. Rev. Inorg. Chem. 2016, 36, 1–41. [Google Scholar] [CrossRef]
- Jensen, M.B.; Pettersson, L.G.M.; Swang, O.; Olsbye, U. CO2 sorption on MgO and CaO surfaces: A comparative quantum chemical cluster study. J. Phys. Chem. B 2005, 109, 16774–16781. [Google Scholar] [CrossRef] [PubMed]
- Loder, A.; Siebenhofer, M.; Lux, S. The reaction kinetics of CO2 methanation on a bifunctional Ni/MgO catalyst. J. Ind. Eng. Chem. 2020, 85, 196–207. [Google Scholar] [CrossRef]
- Kleiber, S.; Loder, A.; Siebenhofer, M.; Böhm, A.; Lux, S. Direct reduction of siderite ore combined with catalytic CO/CO2 hydrogenation to methane and methanol: A technology concept. Chem. Ing. Tech. 2022, 94, 701–711. [Google Scholar] [CrossRef]
- Baldauf-Sommerbauer, G.; Lux, S.; Aniser, W.; Bitschnau, B.; Letofsky-Papst, I.; Siebenhofer, M. Steady-state and controlled heating rate methanation of CO2 on Ni/MgO in a bench-scale fixed bed tubular reactor. J. CO2 Util. 2018, 23, 1–9. [Google Scholar] [CrossRef]
- Bowles, J.F.W. (Ed.) Encyclopedia of Geology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2021; ISBN 9780081029091. [Google Scholar]
- Seeger, M.; Otto, W.; Flick, W.; Bickelhaupt, F.; Akkerman, O.S. Magnesium compounds. Ullmann’s Encycl. Ind. Chem. 2011. [Google Scholar] [CrossRef]
- Amodeo, J.; Merkel, S.; Tromas, C.; Carrez, P.; Korte-Kerzel, S.; Cordier, P.; Chevalier, J. Dislocations and plastic deformation in MgO crystals: A review. Crystals 2018, 8, 240. [Google Scholar] [CrossRef] [Green Version]
- European Commission. Best Available Techniques (BAT) Reference Document for the Production of Cement, Lime and Magnesium Oxide, Industrial Emissions Directive 2010/75/EU. Available online: https://eippcb.jrc.ec.europa.eu/sites/default/files/2019-11/CLM_Published_def_0.pdf (accessed on 21 March 2023).
- Baldauf-Sommerbauer, G.; Lux, S.; Aniser, W.; Siebenhofer, M. Reductive Calcination of Mineral Magnesite: Hydrogenation of Carbon Dioxide without Catalysts. Chem. Eng. Technol. 2016, 39, 2035–2041. [Google Scholar] [CrossRef]
- Védrine, J.C. (Ed.) Metal Oxides in Heterogeneous Catalysis; Elsevier: Paris, France, 2018. [Google Scholar]
- Di Cosimo, J.I.; Díez, V.K.; Ferretti, C.; Apesteguía, C.R. Basic Catalysis on MgO: Generation, Characterization and Catalytic Properties of Active Sites; Royal Society of Chemistry: London, UK, 2014. [Google Scholar]
- Wu, M.-C.; Goodman, D.W. Acid/base properties of MgO studied by high resolution electron energy loss spectroscopy. Catal. Lett. 1992, 15, 1–11. [Google Scholar] [CrossRef]
- Kumar, S.; Saxena, S.K. A comparative study of CO2 sorption properties for different oxides. Mater. Renew. Sustain. Energy 2014, 3, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Abedin, A.H.; Rosen, M.A. A critical review of thermochemical energy storage systems. Open Renew. Energy J. 2011, 4, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Yu, C.; Lu, J.; Wei, X.; Wang, W.; Pan, G. Enhanced CO2 adsorption of MgO with alkali metal nitrates and carbonates. Appl. Energy 2020, 263, 114681. [Google Scholar] [CrossRef]
- Han, X.; Wang, L.; Ling, H.; Ge, Z.; Lin, X.; Dai, X.; Chen, H. Critical review of thermochemical energy storage systems based on cobalt, manganese, and copper oxides. Renew. Sustain. Energy Rev. 2022, 158, 112076. [Google Scholar] [CrossRef]
- Kyaw, K.; Matsuda, H.; Hasatani, M. Applicability of carbonation/decarbonation reactions to high-temperature thermal energy storage and temperature upgrading. J. Chem. Eng. Japan 1996, 29, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Pardo, P.; Deydier, A.; Anxionnaz-Minvielle, Z.; Rougé, S.; Cabassud, M.; Cognet, P. A review on high temperature thermochemical heat energy storage. Renew. Sustain. Energy Rev. 2014, 32, 591–610. [Google Scholar] [CrossRef] [Green Version]
- Müller, D.; Knoll, C.; Gravogl, G.; Artner, W.; Werner, A.; Welch, J.M.; Harasek, M.; Miletich, R.; Weinberger, P. Low-temperature carbonatization of metal oxides. Energy Procedia 2019, 158, 4870–4881. [Google Scholar] [CrossRef]
- Shkatulov, A.; Miura, H.; Kim, S.T.; Zamengo, M.; Harada, T.; Takasu, H.; Kato, Y.; Aristov, Y. Thermochemical storage of medium-temperature heat using MgO promoted with eutectic ternary mixture LiNO3-NaNO3-KNO3. J. Energy Storage 2022, 51, 104409. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, Y.; Guo, Y.; Bai, S.; Song, M.; Huang, P.; Wei, X.; Sun, J.; Li, C.; Zhang, J.; et al. Alkali metal nitrates promoted MgO composites with high CO2 uptake for thermochemical energy storage. ACS Appl. Energy Mater. 2021, 4, 9513–9524. [Google Scholar] [CrossRef]
- Flegkas, S.; Birkelbach, F.; Winter, F.; Groenewold, H.; Werner, A. Profitability analysis and capital cost estimation of a thermochemical energy storage system utilizing fluidized bed reactors and the reaction system MgO/Mg(OH)2. Energies 2019, 12, 4788. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Zhou, T.; Gao, Y.; Louis, B.; O’Hare, D.; Wang, Q. Molten salts-modified MgO-based adsorbents for intermediate-temperature CO2 capture: A review. J. Energy Chem. 2017, 26, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Hiremath, V.; Shavi, R.; Seo, J.G. Controlled oxidation state of Ti in MgO-TiO2 composite for CO2 capture. Chem. Eng. J. 2017, 308, 177–183. [Google Scholar] [CrossRef]
- Ruhaimi, A.H.; Aziz, M.; Jalil, A.A. Magnesium oxide-based adsorbents for carbon dioxide capture: Current progress and future opportunities. J. CO2 Util. 2021, 43, 101357. [Google Scholar] [CrossRef]
- Wang, Q.; Luo, J.; Zhong, Z.; Borgna, A. CO2 capture by solid adsorbents and their applications: Current status and new trends. Energy Environ. Sci. 2011, 4, 42–55. [Google Scholar] [CrossRef]
- Meis, N.N.A.H.; Bitter, J.H.; de Jong, K.P. Support and size effects of activated hydrotalcites for precombustion CO2 capture. Ind. Eng. Chem. Res. 2010, 49, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, Y.; Takaoka, K.; Yamabe, S.; Ito, T. Interaction of CO2 with magnesium oxide surfaces: A TPD, FTIR, and cluster-model calculation study. J. Phys. Chem. 1995, 99, 3704–3710. [Google Scholar] [CrossRef]
- Huang, J.; Li, X.; Wang, X.; Fang, X.; Wang, H.; Xu, X. New insights into CO2 methanation mechanisms on Ni/MgO catalysts by DFT calculations: Elucidating Ni and MgO roles and support effects. J. CO2 Util. 2019, 33, 55–63. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.M.; Park, J.-N. Bifunctional mechanism of CO2 methanation on Pd-MgO/SiO2 catalyst: Independent roles of MgO and Pd on CO2 methanation. J. Phys. Chem. C 2010, 114, 7128–7131. [Google Scholar] [CrossRef]
- Falsig, H.; Hvolbæk, B.; Kristensen, I.S.; Jiang, T.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. Trends in the catalytic CO oxidation activity of nanoparticles. Angew. Chem. 2008, 120, 4913–4917. [Google Scholar] [CrossRef]
- Moses, P.G.; Hinnemann, B.; Topsøe, H.; Nørskov, J.K. The effect of Co-promotion on MoS2 catalysts for hydrodesulfurization of thiophene: A density functional study. J. Catal. 2009, 268, 201–208. [Google Scholar] [CrossRef]
- Lauritsen, J.V.; Kibsgaard, J.; Helveg, S.; Topsøe, H.; Clausen, B.S.; Laegsgaard, E.; Besenbacher, F. Size-dependent structure of MoS2 nanocrystals. Nat. Nanotechnol. 2007, 2, 53–58. [Google Scholar] [CrossRef]
- Gao, L.Z.; Au, C.T. CO2 hydrogenation to methanol on a YBa2Cu3O7 catalyst. J. Catal. 2000, 189, 1–15. [Google Scholar] [CrossRef]
- Causa, M.; Dovesi, R.; Kotomin, E.; Pisani, C. The MgO(110) surface and CO adsorption thereon. I. Clean (110) surface. J. Phys. C Solid State Phys. 1987, 20, 4983–4990. [Google Scholar] [CrossRef]
- Usseinov, A.B.; Akilbekov, A.T.; Kotomin, E.A.; Popov, A.I.; Seitov, D.D.; Nekrasov, K.A.; Giniyatova, S.G.; Karipbayev, Z.T. The first principles calculations of CO2 adsorption on (1010) ZnO surface. In Proceedings of the Physics, Technologies and Innovation (Pti-2019): Proceedings of the Vi International Young Researchers’ Conference, Ekaterinburg, Russia, 20–23 May 2019; p. 20181. [Google Scholar]
- Kantorovich, L.N.; Shluger, A.L.; Gillan, M.J. What Can We Learn About Perfect and Defective MgO (001) Surface Using Density Functional Theory? In Defects and Surface-Induced Effects in Advanced Perovskites; Borstel, G., Krumins, A., Millers, D., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 49–60. ISBN 978-94-011-4030-0. [Google Scholar]
- Yu, H.; Wang, X.; Shu, Z.; Fujii, M.; Song, C. Al2O3 and CeO2-promoted MgO sorbents for CO2 capture at moderate temperatures. Front. Chem. Sci. Eng. 2018, 12, 83–93. [Google Scholar] [CrossRef]
- Jin, S.; Bang, G.; Liu, L.; Lee, C.-H. Synthesis of mesoporous MgO–CeO2 composites with enhanced CO2 capture rate via controlled combustion. Microporous Mesoporous Mater. 2019, 288, 109587. [Google Scholar] [CrossRef]
- Jin, S.; Ko, K.-J.; Song, Y.-G.; Lee, K.; Lee, C.-H. Fabrication and kinetic study of spherical MgO agglomerates via water-in-oil method for pre-combustion CO2 capture. Chem. Eng. J. 2019, 359, 285–297. [Google Scholar] [CrossRef]
- Gao, W.; Zhou, T.; Gao, Y.; Wang, Q.; Lin, W. Study on MNO3/NO2 (M = Li, Na, and K)/MgO composites for intermediate-temperature CO2 capture. Energy Fuels 2019, 33, 1704–1712. [Google Scholar] [CrossRef]
- Chen, J.L.; Dong, X.Y.M.; Shi, C.L.; Li, S.H.; Wang, Y.; Zhu, J.H. Fabrication of strong solid base FeO–MgO for warm CO2 capture. Clean—Soil Air Water 2019, 47, 1800447. [Google Scholar] [CrossRef]
- Li, P.; Chen, R.; Lin, Y.; Li, W. General approach to facile synthesis of MgO-based porous ultrathin nanosheets enabling high-efficiency CO2 capture. Chem. Eng. J. 2021, 404, 126459. [Google Scholar] [CrossRef]
- Wang, J.; Huang, L.; Yang, R.; Zhang, Z.; Wu, J.; Gao, Y.; Wang, Q.; O’Hare, D.; Zhong, Z. Recent advances in solid sorbents for CO2 capture and new development trends. Energy Environ. Sci. 2014, 7, 3478–3518. [Google Scholar] [CrossRef]
- Xiao, G.; Singh, R.; Chaffee, A.; Webley, P. Advanced adsorbents based on MgO and K2CO3 for capture of CO2 at elevated temperatures. Int. J. Greenh. Gas Control 2011, 5, 634–639. [Google Scholar] [CrossRef]
- Ito, T. Initial sintering of magnesium oxide in carbon dioxide. J. Chem. Soc. Faraday Trans. 1982, 1, 1603–1613. [Google Scholar] [CrossRef]
- Ho, K.; Jin, S.; Zhong, M.; Vu, A.-T.; Lee, C.-H. Sorption capacity and stability of mesoporous magnesium oxide in post-combustion CO2 capture. Mater. Chem. Phys. 2017, 198, 154–161. [Google Scholar] [CrossRef]
- Vu, A.-T.; Ho, K.; Jin, S.; Lee, C.-H. Double sodium salt-promoted mesoporous MgO sorbent with high CO2 sorption capacity at intermediate temperatures under dry and wet conditions. Chem. Eng. J. 2016, 291, 161–173. [Google Scholar] [CrossRef]
- Hiremath, V.; Shavi, R.; Gil Seo, J. Mesoporous magnesium oxide nanoparticles derived via complexation-combustion for enhanced performance in carbon dioxide capture. J. Colloid Interface Sci. 2017, 498, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Tuan, V.A.; Lee, C.H. Preparation of rod-like MgO by simple precipitation method for CO2 capture at ambient temperature. Vietnam. J. Chem. 2018, 56, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Zhou, T.; Wang, Q. Controlled synthesis of MgO with diverse basic sites and its CO2 capture mechanism under different adsorption conditions. Chem. Eng. J. 2018, 336, 710–720. [Google Scholar] [CrossRef]
- Chen, A.; Yu, Y.; Li, Y.; Li, Y.; Jia, M. Solid-state grinding synthesis of ordered mesoporous MgO/carbon spheres composites for CO2 capture. Mater. Lett. 2016, 164, 520–523. [Google Scholar] [CrossRef]
- Park, J.-N.; McFarland, E.W. A highly dispersed Pd–Mg/SiO2 catalyst active for methanation of CO2. J. Catal. 2009, 266, 92–97. [Google Scholar] [CrossRef]
- Fukuda, Y.; Tanabe, K. Infrared study of carbon dioxide adsorbed on magnesium and calcium oxides. BCSJ 1973, 46, 1616–1619. [Google Scholar] [CrossRef] [Green Version]
- Ahn, C.-I.; Jeong, D.-W.; Cho, J.M.; Na, H.-S.; Jang, W.-J.; Roh, H.-S.; Choi, J.-H.; Um, S.H.; Bae, J.W. Water gas shift reaction on the Mn-modified ordered mesoporous Co3O4. Microporous Mesoporous Mater. 2016, 221, 204–211. [Google Scholar] [CrossRef]
- Highfield, J.; Bu, J.; Fagerlund, J.; Zevenhoven, R. The promoter effect of steam in gas-solid CO2 mineralisation. In Proceedings of the Conference: 11th International Conference on Carbon Dioxide Utilization (ICCDU XI), Dijon, France, 27–30 June 2011. [Google Scholar]
- Ding, Y.-D.; Song, G.; Liao, Q.; Zhu, X.; Chen, R. Bench scale study of CO2 adsorption performance of MgO in the presence of water vapor. Energy 2016, 112, 101–110. [Google Scholar] [CrossRef]
- Ram Reddy, M.K.; Xu, Z.P.; Lu, G.Q.; Da Diniz Costa, J.C. Influence of water on high-temperature CO2 capture using layered double hydroxide derivatives. Ind. Eng. Chem. Res. 2008, 47, 2630–2635. [Google Scholar] [CrossRef]
- Duan, Y.; Sorescu, D.C. CO2 capture properties of alkaline earth metal oxides and hydroxides: A combined density functional theory and lattice phonon dynamics study. J. Chem. Phys. 2010, 133, 74508. [Google Scholar] [CrossRef]
- Fagerlund, J.; Highfield, J.; Zevenhoven, R. Kinetics studies on wet and dry gas–solid carbonation of MgO and Mg(OH)2 for CO2 sequestration. RSC Adv. 2012, 2, 10380. [Google Scholar] [CrossRef]
- Baldauf-Sommerbauer, G.; Lux, S.; Aniser, W.; Siebenhofer, M. Synthesis of carbon monoxide from hydrogen and magnesite/dolomite. Chem. Ing. Tech. 2017, 89, 172–179. [Google Scholar] [CrossRef]
- Smith, H.J.; Fahrenkamp-Uppenbrink, J.; Coontz, R. Carbon capture and sequestration. Clearing the air. Introduction. Science 2009, 325, 1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, S. Carbon capture and sequestration. Science 2009, 325, 1599. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-E.; Yoo, J.S. New CO2 chemistry—Recent advances in utilizing CO2 as an oxidant and current understanding on its role. In Carbon Dioxide Utilization for Global Sustainability, Proceedings of 7th the International Conference on Carbon Dioxide Utilization; Elsevier: Amsterdam, The Netherlands, 2004; pp. 303–314. ISBN 9780444516008. [Google Scholar]
- Steeneveldt, R.; Berger, B.; Torp, T.A. CO2 capture and storage. Chem. Eng. Res. Des. 2006, 84, 739–763. [Google Scholar] [CrossRef]
- Li, L.; King, D.L.; Nie, Z.; Howard, C. Magnesia-stabilized calcium oxide absorbents with improved durability for high temperature CO2 capture. Ind. Eng. Chem. Res. 2009, 48, 10604–10613. [Google Scholar] [CrossRef]
- Manovic, V.; Anthony, E.J. CaO-based pellets supported by calcium aluminate cements for high-temperature CO2 capture. Environ. Sci. Technol. 2009, 43, 7117–7122. [Google Scholar] [CrossRef]
- Sandru, M.; Haukebø, S.H.; Hägg, M.-B. Composite hollow fiber membranes for CO2 capture. J. Membr. Sci. 2010, 346, 172–186. [Google Scholar] [CrossRef]
- Bachu, S. CO2 storage in geological media: Role, means, status and barriers to deployment. Prog. Energy Combust. Sci. 2008, 34, 254–273. [Google Scholar] [CrossRef]
- Haszeldine, R.S. Carbon capture and storage: How green can black be? Science 2009, 325, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Xu, J.; Liang, B.; Duan, H.; Hou, B.; Huang, Y. Catalytic carbon dioxide hydrogenation to methane: A review of recent studies. J. Energy Chem. 2016, 25, 553–565. [Google Scholar] [CrossRef]
- Guo, M.; Lu, G. The effect of impregnation strategy on structural characters and CO2 methanation properties over MgO modified Ni/SiO2 catalysts. Catal. Commun. 2014, 54, 55–60. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Treviño, M.A.; Farrauto, R.J. CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Varun, Y.; Sreedhar, I.; Singh, S.A. Highly stable M/NiO–MgO (M = Co, Cu and Fe) catalysts towards CO2 methanation. Int. J. Hydrog. Energy 2020, 45, 28716–28731. [Google Scholar] [CrossRef]
- Fan, M.T.; Lin, J.D.; Zhang, H.B.; Liao, D.W. In situ growth of carbon nanotubes on Ni/MgO: A facile preparation of efficient catalysts for the production of synthetic natural gas from syngas. Chem. Commun. 2015, 51, 15720–15723. [Google Scholar] [CrossRef]
- Nakayama, T.; Ichikuni, N.; Sato, S.; Nozaki, F. Ni/MgO catalyst prepared using citric acid for hydrogenation of carbon dioxide. Appl. Catal. A Gen. 1997, 158, 185–199. [Google Scholar] [CrossRef]
- Takezawa, N.; Terunuma, H.; Shimokawabe, M.; Kobayashib, H. Methanation of carbon dioxide: Preparation of Ni/MgO catalysts and their performance. Appl. Catal. 1986, 23, 291–298. [Google Scholar] [CrossRef]
- Bette, N.; Thielemann, J.; Schreiner, M.; Mertens, F. Methanation of CO2 over a (Mg,Al)Ox Supported Nickel Catalyst Derived from a (Ni,Mg,Al)-Hydrotalcite-like Precursor. ChemCatChem 2016, 8, 2903–2906. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhang, X.; Rui, N.; Hu, X.; Liu, C. Structural effect of Ni/ZrO2 catalyst on CO2 methanation with enhanced activity. Appl. Catal. B Environ. 2019, 244, 159–169. [Google Scholar] [CrossRef]
- Pan, Q.; Peng, J.; Sun, T.; Wang, S.; Wang, S. Insight into the reaction route of CO2 methanation: Promotion effect of medium basic sites. Catal. Commun. 2014, 45, 74–78. [Google Scholar] [CrossRef]
- Westermann, A.; Azambre, B.; Bacariza, M.C.; Graça, I.; Ribeiro, M.F.; Lopes, J.M.; Henriques, C. Insight into CO2 methanation mechanism over NiUSY zeolites: An operando IR study. Appl. Catal. B Environ. 2015, 174–175, 120–125. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; He, Z.; Libby, M.C.; Farrauto, R.J. Catalysts and adsorbents for CO2 capture and conversion with dual function materials: Limitations of Ni-containing DFMs for flue gas applications. J. CO2 Util. 2019, 31, 143–151. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, Y.; Li, J.; Wei, S.; Gao, X.; Wang, P. Combustion-impregnation preparation of Ni/SiO2 catalyst with improved low-temperature activity for CO2 methanation. Int. J. Hydrog. Energy 2021, 46, 20919–20929. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, C.; Gao, P.; Wang, H.; Li, X.; Zhong, L.; Wei, W.; Sun, Y. A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons. Catal. Sci. Technol. 2017, 7, 4580–4598. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Ban, H.; Li, C.; Asami, K.; Fujimoto, K. Influence of rare-earth elements (La, Ce, Nd and Pr) on the performance of Cu/Zn/Zr catalyst for CH3OH synthesis from CO2. Catal. Commun. 2014, 54, 50–54. [Google Scholar] [CrossRef]
- Guo, X.; Mao, D.; Lu, G.; Wang, S.; Wu, G. Glycine–nitrate combustion synthesis of CuO–ZnO–ZrO2 catalysts for methanol synthesis from CO2 hydrogenation. J. Catal. 2010, 271, 178–185. [Google Scholar] [CrossRef]
- Hu, B.; Yin, Y.; Liu, G.; Chen, S.; Hong, X.; Tsang, S.C.E. Hydrogen spillover enabled active Cu sites for methanol synthesis from CO2 hydrogenation over Pd doped CuZn catalysts. J. Catal. 2018, 359, 17–26. [Google Scholar] [CrossRef]
- Fujitani, T.; Saito, M.; Kanai, Y.; Kakumoto, T.; Watanabe, T.; Nakamura, J.; Uchijima, T. The role of metal oxides in promoting a copper catalyst for methanol synthesis. Catal. Lett. 1994, 25, 271–276. [Google Scholar] [CrossRef]
- Baltes, C.; Vukojevic, S.; Schuth, F. Correlations between synthesis, precursor, and catalyst structure and activity of a large set of CuO/ZnO/Al2O3 catalysts for methanol synthesis. J. Catal. 2008, 258, 334–344. [Google Scholar] [CrossRef]
- Robbins, J.L.; Iglesia, E.; Kelkar, C.P.; DeRites, B. Methanol synthesis over Cu/SiO2 catalysts. Catal. Lett. 1991, 10, 1–10. [Google Scholar] [CrossRef]
- Ren, H.; Xu, C.-H.; Zhao, H.-Y.; Wang, Y.-X.; Liu, J.; Liu, J.-Y. Methanol synthesis from CO2 hydrogenation over Cu/γ-Al2O3 catalysts modified by ZnO, ZrO2 and MgO. J. Ind. Eng. Chem. 2015, 28, 261–267. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Neja, S.Š.; Blaž, L. Correlation between synthesis pH, structure and Cu/MgO/Al2O3 heterogeneous catalyst activity and selectivity in CO2 hydrogenation to methanol. J. CO2 Util. 2018, 28, 189–199. [Google Scholar] [CrossRef]
- Zhan, H.; Li, F.; Gao, P.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Methanol synthesis from CO2 hydrogenation over La–M–Cu–Zn–O (M = Y, Ce, Mg, Zr) catalysts derived from perovskite-type precursors. J. Power Sources 2014, 251, 113–121. [Google Scholar] [CrossRef]
- Liu, C.; Guo, X.; Guo, Q.; Mao, D.; Yu, J.; Lu, G. Methanol synthesis from CO2 hydrogenation over copper catalysts supported on MgO-modified TiO2. J. Mol. Catal. A Chem. 2016, 425, 86–93. [Google Scholar] [CrossRef]
- Zander, S.; Kunkes, E.L.; Schuster, M.E.; Schumann, J.; Weinberg, G.; Teschner, D.; Jacobsen, N.; Schlögl, R.; Behrens, M. The role of the oxide component in the development of copper composite catalysts for methanol synthesis. Angew. Chem. Int. Ed. Engl. 2013, 52, 6536–6540. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, Y.; Iwama, Y.; Tsubaki, N. Mechanistic study of a new low-temperature methanol synthesis on Cu/MgO catalysts. Appl. Catal. A Gen. 2005, 288, 126–133. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.; Wei, W.; Sun, Y. Solid base and their performance in synthesis of propylene glycol methyl ether. J. Mol. Catal. A Chem. 2005, 231, 83–88. [Google Scholar] [CrossRef]
- Madeira, L.M.; Martín-Aranda, R.M.; Maldonado-Hódar, F.J.; Fierro, J.; Portela, M.F. Oxidative dehydrogenation of n-butane over alkali and alkaline earth-promoted α-NiMoO4 catalysts. J. Catal. 1997, 169, 469–479. [Google Scholar] [CrossRef]
- Dasireddy, V.D.; Štefančič, N.S.; Huš, M.; Likozar, B. Effect of alkaline earth metal oxide (MO) Cu/MO/Al2O3 catalysts on methanol synthesis activity and selectivity via CO2 reduction. Fuel 2018, 233, 103–112. [Google Scholar] [CrossRef]
- Fichtl, M.B.; Schumann, J.; Kasatkin, I.; Jacobsen, N.; Behrens, M.; Schlögl, R.; Muhler, M.; Hinrichsen, O. Counting of oxygen defects versus metal surface sites in methanol synthesis catalysts by different probe molecules. Angew. Chem. Int. Ed. Engl. 2014, 53, 7043–7047. [Google Scholar] [CrossRef]
- Niu, J.; Liu, H.; Jin, Y.; Fan, B.; Qi, W.; Ran, J. Comprehensive review of Cu-based CO2 hydrogenation to CH3OH: Insights from experimental work and theoretical analysis. Int. J. Hydrog. Energy 2022, 47, 9183–9200. [Google Scholar] [CrossRef]
- Cao, A.; Wang, Z.; Li, H.; Elnabawy, A.O.; Nørskov, J.K. New insights on CO and CO2 hydrogenation for methanol synthesis: The key role of adsorbate-adsorbate interactions on Cu and the highly active MgO-Cu interface. J. Catal. 2021, 400, 325–331. [Google Scholar] [CrossRef]
- Kleiber, S.; Pallua, M.; Siebenhofer, M.; Lux, S. Catalytic hydrogenation of CO2 to methanol over Cu/MgO catalysts in a semi-continuous reactor. Energies 2021, 14, 4319. [Google Scholar] [CrossRef]
- Grabow, L.C.; Mavrikakis, M. Mechanism of methanol synthesis on Cu through CO2 and CO hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Lam, E.; Corral-Pérez, J.J.; Larmier, K.; Noh, G.; Wolf, P.; Comas-Vives, A.; Urakawa, A.; Copéret, C. CO2 hydrogenation on Cu/Al2O3: Role of the metal/support Interface in driving activity and selectivity of a bifunctional catalyst. Angew. Chem. Int. Ed. Engl. 2019, 58, 13989–13996. [Google Scholar] [CrossRef]
- Poto, S.; van Vico Berkel, D.; Gallucci, F.; Fernanda Neira d’Angelo, M. Kinetic modelling of the methanol synthesis from CO2 and H2 over a CuO/CeO2/ZrO2 catalyst: The role of CO2 and CO hydrogenation. Chem. Eng. J. 2022, 435, 134946. [Google Scholar] [CrossRef]
- Tabatabaei, J.; Sakakini, B.H.; Waugh, K.C. On the mechanism of methanol synthesis and the water-gas shift reaction on ZnO. Catal. Lett. 2006, 110, 77–84. [Google Scholar] [CrossRef]
- Shido, T.; Iwasawa, Y. The effect of coadsorbates in reverse water-gas shift reaction on ZnO, in relation to reactant-promoted reaction mechanism. J. Catal. 1993, 140, 575–584. [Google Scholar] [CrossRef]





| Catalyst Composition | Preparation Method | Operation Conditions | Performance | Comments | Ref. |
|---|---|---|---|---|---|
| Ni/MgO (wNi = 0–27 wt%) | wet impregnation | T = 533–648 K GHSV = 3.7 m3 kg−1 h−1 v(H2):v(CO2):v(N2) = 4:1:5 | XCO2 = 87% YCH4 = 99% | [13] | |
| Ni/MgO (wNi = 11 and 17 wt%) | wetimpregnation | T = 603 K GHSV = 1.24–5 m3 kg−1 h−1 | XCO2 = 70% YCH4 = 99 % | [15] | |
| NiO/MgO (with and without impregnation with 2% Co, Cu, Fe) | sonochemicalmethod | T = 673 K, GHSV = 47.76 h−1 | XCO2 = 85% YCH4 = 98% (XCO2 = 90% (Co), 86% (Cu), 89% (Fe) YCH4 = 99% (Co), 94% (Cu), 96% (Fe)) | [85] | |
| Ni/MgO-CNTs 1 | precipitation | T = 473–713 K GHSV = 40 m3 kg−1 h−1 v(H2):v(CO):v(N2):v(CO2) = 75:15:5:5 | X(CO+CO2) = ~100% YCH4 = ~100% | [86] | |
| Ni/MgO (wNi = 30–90 wt%) | citric acid complex method | T = 553 K v(CO2):v(H2) = 1:8 | XCO2 = 62–85% SCH4 = 99–100% | space–time yield of CH4: 8.7–28.2 min−1 | [87] |
| Ni/MgO (wNi = 70 w%t) | coprecipitation | T = 553 K v(CO2):v(H2) = 1:8 | XCO2 = 32% SCH4 = 68% | space–time yield of CH4: 2.7 min−1 | [87] |
| Mg-Al-CO3 LDH 2 catalyst | coprecipitation | T = 473–573 K v(CO2):v(O2):v(N2) = 14:4:82 | CO2 sorption: 2.72% (dry sorption), 3.14% (wet condition, 12% water) | [69] | |
| Ni/MgO Ni(111), MgO (110) | DFT calculations | [40] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suksumrit, K.; Kleiber, S.; Lux, S. The Role of Carbonate Formation during CO2 Hydrogenation over MgO-Supported Catalysts: A Review on Methane and Methanol Synthesis. Energies 2023, 16, 2973. https://doi.org/10.3390/en16072973
Suksumrit K, Kleiber S, Lux S. The Role of Carbonate Formation during CO2 Hydrogenation over MgO-Supported Catalysts: A Review on Methane and Methanol Synthesis. Energies. 2023; 16(7):2973. https://doi.org/10.3390/en16072973
Chicago/Turabian StyleSuksumrit, Kamonrat, Sascha Kleiber, and Susanne Lux. 2023. "The Role of Carbonate Formation during CO2 Hydrogenation over MgO-Supported Catalysts: A Review on Methane and Methanol Synthesis" Energies 16, no. 7: 2973. https://doi.org/10.3390/en16072973
APA StyleSuksumrit, K., Kleiber, S., & Lux, S. (2023). The Role of Carbonate Formation during CO2 Hydrogenation over MgO-Supported Catalysts: A Review on Methane and Methanol Synthesis. Energies, 16(7), 2973. https://doi.org/10.3390/en16072973