Abstract
Hydrogen storage materials play a pivotal role in the development of a sustainable hydrogen economy. However, the discovery and optimization of high-performance storage materials remain a significant challenge due to the complex interplay of structural, thermodynamic and kinetic factors. Computational materials science has emerged as a powerful tool to accelerate the design and development of novel hydrogen storage materials by providing atomic-level insights into the storage mechanisms and guiding experimental efforts. In this comprehensive review, we discuss the recent advances in crystal structure prediction and performance assessment of hydrogen storage materials from a computational perspective. We highlight the applications of state-of-the-art computational methods, including density functional theory (DFT), molecular dynamics (MD) simulations, and machine learning (ML) techniques, in screening, evaluating, and optimizing storage materials. Special emphasis is placed on the prediction of stable crystal structures, assessment of thermodynamic and kinetic properties, and high-throughput screening of material space. Furthermore, we discuss the importance of multiscale modeling approaches that bridge different length and time scales, providing a holistic understanding of the storage processes. The synergistic integration of computational and experimental studies is also highlighted, with a focus on experimental validation and collaborative material discovery. Finally, we present an outlook on the future directions of computationally driven materials design for hydrogen storage applications, discussing the challenges, opportunities, and strategies for accelerating the development of high-performance storage materials. This review aims to provide a comprehensive and up-to-date account of the field, stimulating further research efforts to leverage computational methods to unlock the full potential of hydrogen storage materials.
1. Introduction
The global energy landscape is undergoing profound changes, driven by the urgent need to mitigate climate change and the increasing demand for clean and sustainable energy sources [1,2]. With governments and businesses worldwide placing growing emphasis on carbon emission reductions, finding viable alternative energy sources has become paramount. Hydrogen, due to its high energy density, clean combustion, and potential for renewable production, has emerged as a promising energy carrier [3,4,5]. Unlike conventional fossil fuels, hydrogen combustion produces only water, with no harmful emissions, making it a crucial pathway for reducing greenhouse gas emissions. Furthermore, by utilizing renewable energy sources such as wind, solar, or hydroelectric power for hydrogen production via water electrolysis, a zero-carbon emission lifecycle from production to usage is achieved, offering a sustainable energy solution [6,7].
However, widespread adoption of hydrogen as a fuel is severely constrained by traditional storage technologies [8,9]. Table 1 summarizes the advantages and disadvantages of various hydrogen storage technologies. High-pressure compression and cryogenic liquefaction, as mainstream methods, efficiently store hydrogen but pose significant challenges in terms of safety, energy efficiency, and infrastructure compatibility [10]. High-pressure storage requires robust and heavy containers, increasing transportation costs and introducing potential safety risks. On the other hand, cryogenic liquefaction consumes substantial energy for hydrogen liquefaction and requires complex insulation technologies to maintain low temperatures, which are economically and practically impractical for large-scale applications [11,12]. In contrast, solid-state hydrogen storage technologies are highly regarded for their potential to store hydrogen at high densities under moderate conditions. These advanced materials not only effectively store hydrogen but also allow controlled hydrogen release in practical operations, meeting requirements for safety, cost-effectiveness, and utility [13,14]. Future developments should further analyze the pros and cons of various hydrogen storage technologies in terms of cost, reliability, safety, and practicality, but solid-state hydrogen storage technology undoubtedly holds the key to transitioning to a hydrogen-based economy.

Table 1.
Comprehensive comparison of various hydrogen storage technologies.
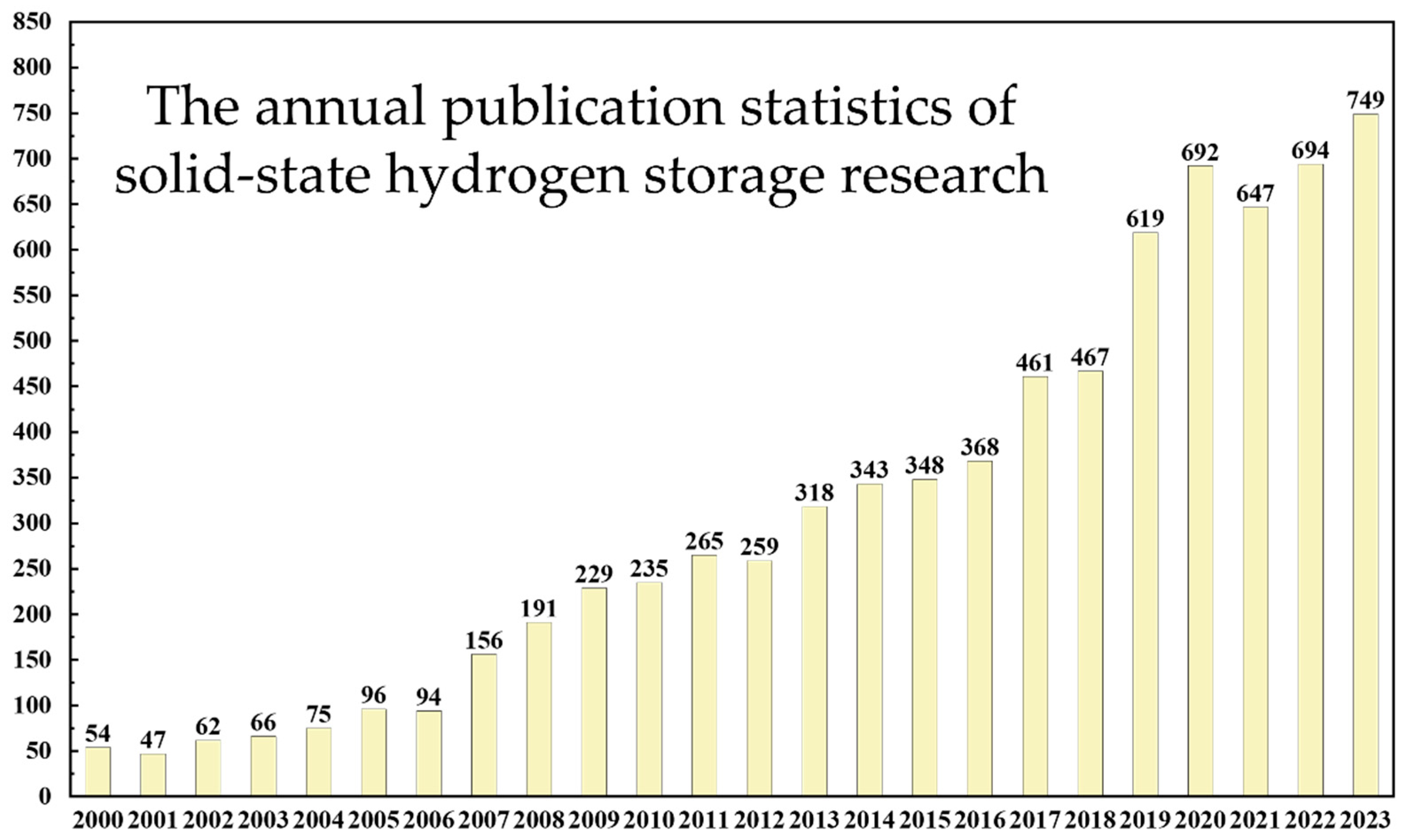
Solid-state hydrogen materials are widely viewed as a promising solution for sustainable energy storage and transmission. Research on solid-state hydrogen materials has seen significant development worldwide, as depicted in Figure 1, which shows an increasing trend in research publications in the field of solid-state hydrogen storage, despite a decline after 2020 due to the pandemic, maintaining an overall growth trajectory. These materials stabilize hydrogen within their structures through chemical reactions or physical adsorption mechanisms, overcoming some limitations of traditional hydrogen storage technologies, such as safety and efficiency issues [15,16]. Major categories include physically adsorptive materials like activated carbon, graphene, and metal–organic frameworks (MOFs), as well as chemically adsorptive ones like metal hydrides and complex hydrides [17,18,19]. Adsorption-based materials like activated carbon and MOFs can rapidly adsorb and release hydrogen at room temperature but may be limited by their hydrogen storage capacity [20,21]. Metal hydrides typically offer high hydrogen storage capacities but face challenges in reaction kinetics and cycling stability, needing solutions for slow reaction rates and thermal decomposition at high temperatures [22,23]. Complex hydrides possess multiphase reaction capabilities and higher hydrogen mass fractions due to their structural complexity but must also overcome periodic thermodynamic reactions to achieve stable hydrogen storage and release [24,25]. These materials often exhibit excellent gas molecule recognition and selectivity but require further enhancement of their adsorption capacity and cycling stability for practical engineering applications [26,27]. Therefore, the development of solid-state hydrogen materials faces challenges in engineering and practicality, requiring innovation in material design, reaction kinetics optimization, and structural engineering to overcome these barriers. Future research will focus on improving the hydrogen storage performance and cycling stability of materials to realize their widespread application and commercialization potential in clean energy technologies.
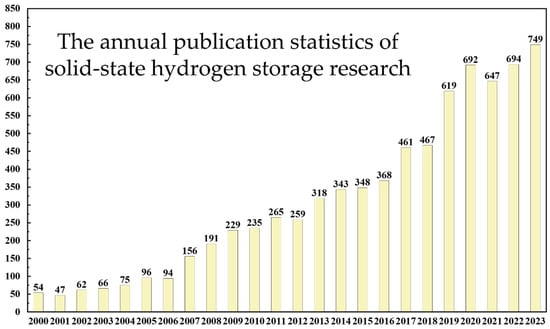
Figure 1.
Statistics on the number of research papers published in the field of solid-state hydrogen storage (as of the end of 2023).
To overcome these challenges and further optimize hydrogen storage technologies, the application of computational materials science becomes crucial. Using advanced computational techniques such as density functional theory (DFT), molecular dynamics simulations (MD), and machine learning (ML), researchers can simulate and predict the structure, stability, and hydrogen storage performance of solid-state hydrogen materials [28,29]. These methods enable scientists to rapidly screen and optimize a large number of candidate materials outside the laboratory, accelerating the discovery and development process of new materials. By understanding materials’ atomic-level behavior at the computational level, researchers can precisely design new materials with ideal hydrogen storage characteristics and guide experimental verification towards the most promising directions [30,31]. Furthermore, the application of machine learning in materials science opens up new possibilities for the design and optimization of hydrogen storage materials [30,31]. By analyzing large volumes of structural and performance data, machine learning models can identify key features and interactions within materials, thereby predicting the most promising candidate materials and speeding up the experimental verification process. This data-driven approach not only enhances the efficiency of material discovery but also uncovers associations and trends that traditional methods may overlook, paving the way for further innovation in hydrogen storage technology [32].
In conclusion, solid-state hydrogen materials, as a crucial component of next-generation hydrogen storage technologies, are poised to advance towards more mature and implementable stages through computation-driven design and optimization. With technological advancements and deepened understanding, these materials hold the promise of providing reliable and sustainable solutions for achieving a hydrogen-based economy, driving the global transition towards a sustainable energy future.
The overall structure of this review is as follows: Section 2 provides an overview of the key computational methods used in hydrogen storage research, including DFT, MD simulations, and ML techniques. Section 3 focuses on the application of these methods in predicting stable crystal structures, assessing thermodynamic and kinetic properties, and high-throughput screening of materials. Section 4 discusses multiscale modeling approaches and their role in bridging different length and time scales. Section 5 highlights the synergistic integration of computational and experimental studies, emphasizing the importance of experimental validation and collaborative materials discovery. Section 6 presents an outlook on the future directions and challenges in the design of computationally driven hydrogen storage materials. Finally, Section 7 concludes the review by summarizing the key points and providing perspectives on the role of computational materials science in advancing hydrogen storage research.
2. Computational Methods for Hydrogen Storage Materials
2.1. Density Functional Theory
Density functional theory (DFT) has become a cornerstone in computational materials science, particularly in the study of hydrogen storage materials [33,34]. DFT, rooted in the fundamental principles of quantum mechanics, offers a powerful approach to understanding and predicting the electronic structure and properties of materials at the atomic level [35]. The foundations of DFT can be traced back to the seminal work of Hohenberg and Kohn [36], who proved that the ground state properties of a many-electron system are uniquely determined by its electron density. This was further developed by Kohn and Sham [37], who introduced a practical scheme for DFT calculations, now known as the Kohn–Sham equations. These developments built upon earlier work in statistical mechanics and quantum theory [38].
In the context of hydrogen storage materials, DFT calculations have been instrumental in predicting stable crystal structures, calculating binding energies, and understanding the electronic properties that influence hydrogen absorption and desorption processes [39,40]. The accuracy and efficiency of DFT make it particularly suitable for screening large numbers of potential storage materials. Recent advancements in DFT methodologies have further improved its applicability to complex systems relevant to hydrogen storage [41]. For instance, the development of van der Waals-corrected functionals has enhanced the accuracy of calculations involving weakly bound systems, which are often encountered in physisorption-based hydrogen storage materials [42]. Additionally, the application of density-functional perturbation theory has enabled more accurate predictions of vibrational properties and phonon spectra, crucial for understanding the thermodynamics of hydrogen storage materials [43].
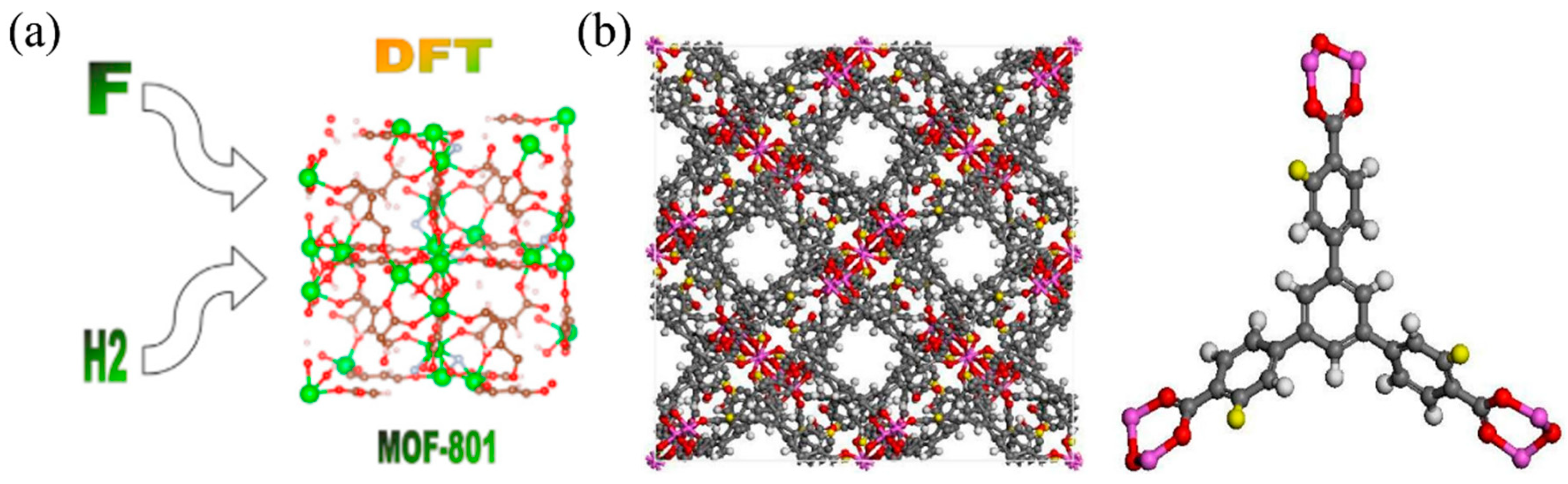
One of the key advantages of DFT is its ability to predict the stable crystal structures of hydrogen storage materials. By exploring the potential energy surface and minimizing the total energy with respect to atomic positions and lattice parameters, the most thermodynamically favorable structures can be identified [44]. This crystal structure prediction capability is particularly valuable for discovering new storage materials and understanding the phase transitions during hydrogen absorption and desorption processes. Zhu et al. [45] employed DFT to compute gas-phase MgHn clusters (n = 1–20), investigating the scientific mechanisms of hydrogen storage by individual Mg atoms. The study revealed that MgH7 and MgH12 gas-phase clusters achieve optimal theoretical sizes for saturation with odd and even numbers of H atoms, respectively. Their hydrogen storage capacities reached 22.69 wt.% and 33.47 wt.%, significantly surpassing MgH2 (7.6 wt.%). Furthermore, two growth modes were identified in these clusters, alternating between odd and even numbers of H atoms, with even-numbered H atoms found to be more stable than odd-numbered ones. Electronic structure studies demonstrated the systematic contributions of Mg and H atoms to molecular orbitals. Through in-depth exploration of atomic charge distribution and topological properties at bond critical points, including bonding pathways, electron density, electron density Laplacian function, electron localization function, and interaction region indicators, a clear and insightful magnesium-hydrogen atomic parity mechanism was established. Figure 2a illustrates the DFT-predicted crystal structure and hydrogen adsorption sites in a metal–organic framework (MOF) material [46]. Similarly, as shown in Figure 2b, six different functional groups were introduced to modify the original MOF-519, resulting in functionalized MOF-519-X (X = NH2, Cl, OH, NO2, F, and CH3). In this simulation, hydrogen atoms on the organic ligand were replaced with functional groups via single bonds [47]. To avoid potential changes in the tetrahedral structure, excessively large substituents were not used to replace hydrogen atoms on the benzene ring. Furthermore, three hydrogen atoms of the same type on the edge benzene ring were replaced with functional groups, yielding functionalized MOF-519 [47]. The ability to visualize the atomic-scale interactions between hydrogen and the host material provides valuable insights into the storage mechanisms and guides the design of improved storage materials.
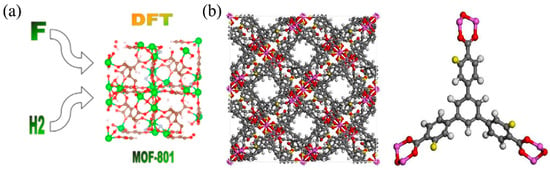
Figure 2.
(a) DFT-predicted crystal structure and hydrogen adsorption sites in a MOF material. Reproduced with permission from [46], Elsevier, 2022. (b) The model of MOF-519-X and structures of organic linker. Atom color scheme: C, gray; H, white; O, red; X, yellow (X = single bond OH, single bond NO2, single bond Cl, single bond NH2, single bond CH3, single bond F). Reproduced with permission from [47], Elsevier, 2019.
Table 2 presents a comparison of the hydrogen binding energies and storage capacities of selected materials predicted by DFT calculations and experimental measurements. The good agreement between the computational and experimental values demonstrates the predictive power of DFT in assessing the hydrogen storage properties of materials.
Table 2.
Comparison of hydrogen binding energies and storage capacities predicted by DFT and experimental measurements for selected materials.
Despite its successes, it’s important to note that the choice of exchange-correlation functional in DFT can significantly impact the results. The accuracy of DFT calculations depends on the choice of exchange-correlation functional, which approximates the complex electron–electron interactions. Commonly used functionals, such as the local density approximation (LDA) and generalized gradient approximation (GGA), may underestimate the binding energies and overestimate the storage capacities, particularly for weakly bound systems [48]. The development of more accurate functionals, such as hybrid functionals and van der Waals-corrected functionals, has helped to mitigate these issues, but at the cost of increased computational expense [49]. Common functionals used in hydrogen storage research include GGA functionals like PBE and hybrid functionals like B3LYP. Each has its strengths and limitations, and the selection often depends on the specific system and properties under investigation [50].
Another limitation of DFT is its scalability to large systems. The computational cost of DFT calculations scales cubically with the number of atoms, making it challenging to study complex materials with large unit cells or disordered structures. However, recent advances in linear-scaling DFT methods and high-performance computing have enabled the simulation of systems containing thousands of atoms, opening new possibilities for studying realistic storage materials [51].
2.2. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations have become an indispensable tool for investigating the dynamic behavior and thermodynamic properties of hydrogen storage materials [52]. MD simulations solve Newton’s equations of motion for a system of interacting particles, allowing the prediction of the time evolution of the system under various conditions. In the context of hydrogen storage, MD simulations have been widely used to study the diffusion, adsorption, and desorption processes of hydrogen in storage materials.
One of the key advantages of MD simulations is their ability to capture the realistic dynamics of hydrogen molecules or atoms within the storage material. By simulating the trajectories of hydrogen and the host material atoms over time, MD simulations can provide insights into the microscopic mechanisms governing the storage process [53]. For example, MD simulations can reveal the preferred diffusion pathways, the role of surface barriers, and the influence of temperature and pressure on the hydrogen uptake and release kinetics.
MD simulations can be performed using either classical force fields or ab initio methods. Classical force fields, such as the reactive force field (ReaxFF) [54] and the universal force field (UFF) [55], describe interatomic interactions using empirical potentials parameterized from experimental data or high-level quantum mechanical calculations. Classical MD simulations are computationally efficient and can handle large systems containing millions of atoms, making them suitable for studying the long-term dynamics and thermodynamic properties of storage materials. Bai et al. [56] analyzed factors influencing H2 density constraints through molecular dynamics simulations. The results confirmed the significant impact of pore size and liquid–wall interactions on hydrogen storage capacity. Specifically, H2 density constraints increased with decreasing pore size only when nanoporous materials met energy requirements. The opposing trends observed below and above the H2 interaction energy threshold were attributed to the presence or absence of adsorbed H2 layers. The study also noted the diminishing benefits of constraints with increasing pressure due to disproportionate changes in adsorption layer density. Akbarzadeh et al. [57] mechanically alloyed MgH2 with 10 wt.% of 50Ce-50Ni and 25Ce-75Ni alloys, producing two distinct types of MgH2–Ce–Ni nanocomposites. Due to Ce’s high brittleness, Ce-containing catalytic compounds effectively reduced the dehydrogenation temperature of MgH2 by forming nanostructured grains. The catalytic effect of Ce–Ni alloys significantly lowered the activation energy of MgH2 from 167 kJ/mol (pure MgH2) to 96 kJ/mol. Moreover, the CN1 catalyst reduced the enthalpy of MgH2 to approximately −50 kJ/mol, aligning closely with results from molecular dynamics simulations.
On the other hand, ab initio MD simulations, also known as first-principles MD or Car–Parrinello MD [58], incorporate the electronic structure calculations on-the-fly during the simulation. By solving the Kohn-Sham equations at each time step, ab initio MD simulations provide a more accurate description of the chemical bonding and reactivity in the system. However, the high computational cost of ab initio MD limits its applicability to smaller systems and shorter time scales compared to classical MD. Table 3 presents a comparison of the hydrogen diffusion coefficients and activation energies obtained from MD simulations and experimental measurements for selected materials. The good agreement between the computational and experimental values demonstrates the capability of MD simulations in predicting the kinetic properties of hydrogen storage materials.
Table 3.
Comparison of hydrogen diffusion coefficients and activation energies obtained from MD simulations and experimental measurements for selected materials.
MD simulations have been instrumental in elucidating the hydrogen storage mechanisms in various materials, such as metal hydrides, complex hydrides, and nanostructured materials. For instance, MD simulations have revealed the importance of surface and subsurface diffusion in the hydrogen absorption and desorption processes of metal nanoparticles [59]. The simulations have shown that the presence of surface defects and strain can significantly enhance the hydrogen diffusion rates and lower the activation barriers, providing valuable insights for the design of nanostructured storage materials. Figure 3 illustrates the MD-simulated diffusion pathways of hydrogen atoms in a magnesium hydride (MgH2) crystal. The visualization of the atomic trajectories helps to understand the microscopic mechanisms of hydrogen transport and identify the rate-limiting steps in the storage process [60].
Figure 3.
MD-simulated diffusion pathways of hydrogen atoms (yellow spheres) in a MgH2 crystal. The magnesium atoms are shown as green spheres. Reproduced with permission from [60], Elsevier, 2019.
Despite the successes of MD simulations in predicting the dynamic behavior of hydrogen storage materials, there are several challenges and limitations to be addressed. The accuracy of classical MD simulations relies on the quality of the force field used to describe the interatomic interactions. Developing reliable and transferable force fields for complex storage materials, particularly those involving chemical reactions and phase transitions, remains a significant challenge. The parameterization of force fields often requires extensive training data from experiments or high-level quantum mechanical calculations, which can be time-consuming and computationally expensive.
Another limitation of MD simulations is the accessibility of time scales. While classical MD simulations can typically reach time scales of nanoseconds to microseconds, many hydrogen storage processes, such as diffusion in bulk materials and long-term cycling stability, occur over much longer time scales. Advanced techniques, such as accelerated MD [61] and kinetic Monte Carlo (KMC) simulations [62], have been developed to overcome the time scale limitation and enable the study of rare events and long-term dynamics.
2.3. Machine Learning Methods
Machine learning (ML) has emerged as a powerful tool for accelerating the discovery and optimization of materials, including those for hydrogen storage applications [63]. ML methods leverage the vast amounts of data generated from experiments and computations to learn patterns and relationships, enabling the prediction of material properties and the identification of promising candidates without the need for extensive experimental or computational testing.
In the context of hydrogen storage, ML has been applied to various tasks, such as predicting the hydrogen adsorption capacities of porous materials, identifying the stable crystal structures of hydrides, and optimizing the composition and synthesis conditions of storage materials [64]. By learning from diverse datasets encompassing structural, thermodynamic, and kinetic properties, ML models can guide the search for novel storage materials with improved performance. Zhou et al. [65] utilized efficient implicit/explicit feature machine learning (ML) to identify key factors influencing capacity, such as MeanIonicChar values and Fe content. Subsequently, targeting fuel cell hydrogenation systems, ML was employed for active feature scanning and composition customization. The results demonstrated that Ti0.9Zr0.12Mn1.2Cr0.55(VFe)0.25 exhibited comprehensive performance (1.90 wt.%/127.30 kg H2/m3) and overwhelming cost-effectiveness compared to existing alloys under moderate temperature and pressure conditions. This further corroborated that ML-driven composition customization avoids substantial experimental investments, providing a novel approach for efficiently acquiring high-performance hydrogen storage materials.
One of the key advantages of ML is its ability to efficiently navigate the vast chemical and structural space of potential storage materials. High-throughput computational screening, such as DFT calculations, can generate large datasets of material properties, but the computational cost becomes prohibitive for exploring all possible combinations of elements and structures. ML models trained on these datasets can predict the properties of new materials with high accuracy and at a fraction of the computational cost, enabling the identification of the most promising candidates for further experimental investigation. Another important application of ML in hydrogen storage research is the development of predictive models for hydrogen adsorption and desorption kinetics. By learning from experimental or computational data on the hydrogen uptake and release rates under different conditions, ML models can predict the kinetic behavior of storage materials and guide the optimization of operating parameters, such as temperature and pressure [66]. Table 4 presents a comparison of the hydrogen adsorption capacities of metal–organic frameworks (MOFs) predicted by ML models and experimental measurements. The high accuracy of the ML predictions demonstrates the potential of ML in accelerating the discovery of high-performance storage materials.
Table 4.
Comparison of hydrogen adsorption capacities of metal–organic frameworks (MOFs) predicted by ML models and experimental measurements.
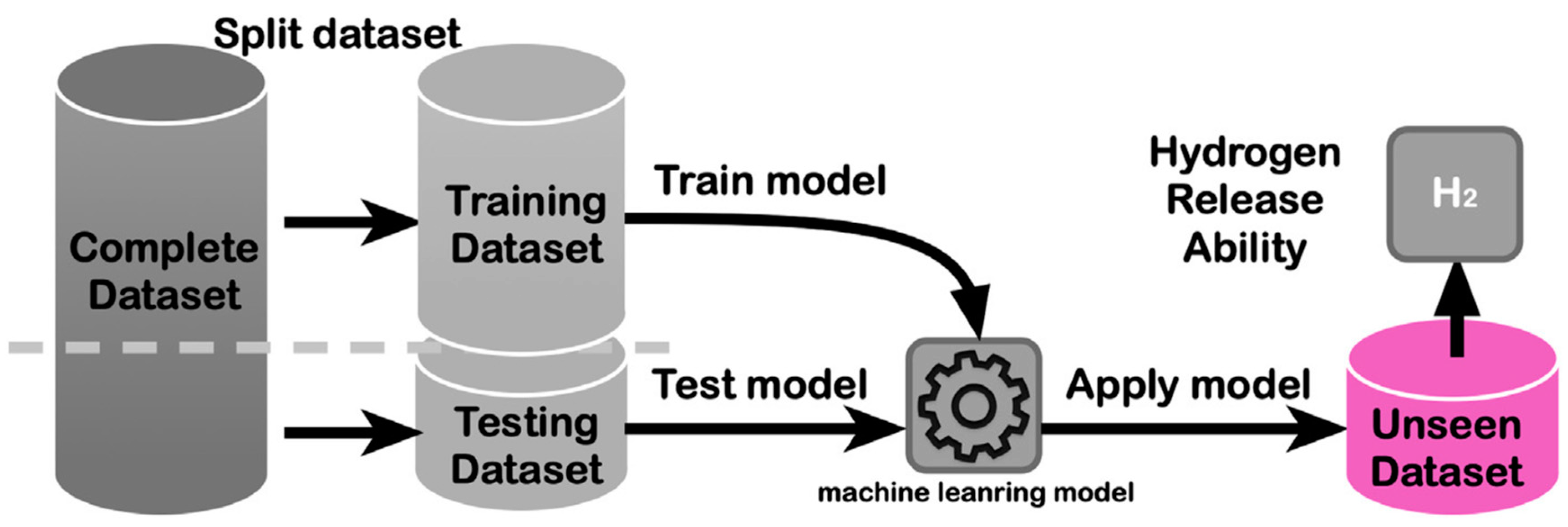
ML methods have also been applied to the prediction of hydrogen diffusion and adsorption mechanisms in storage materials. By analyzing the atomic configurations and trajectories obtained from MD simulations, ML models can learn the underlying patterns and identify the key factors influencing hydrogen mobility and adsorption behavior [67]. This knowledge can guide the rational design of storage materials with enhanced kinetics and storage capacities. Figure 4 shows a schematic representation of a ML workflow for predicting the hydrogen storage properties of materials. The ML model is trained on a dataset of known materials and their properties and then used to predict the properties of new materials. The predictions guide the experimental synthesis and testing of the most promising candidates, leading to the discovery of novel storage materials [68]. Building upon this foundation, the introduction of deep learning algorithms such as multi-layer neural networks enables the capture of intricate patterns and correlations within complex datasets of materials and their properties. This capability facilitates more accurate predictions of material performance based on training data. Furthermore, deep learning models are adept at efficiently processing large volumes of data, which is advantageous in handling diverse material datasets. They excel at learning hierarchical representations of data, revealing subtle relationships that traditional machine learning methods may overlook. By harnessing deep learning in materials science, researchers can expedite the discovery of novel storage materials. The predictions generated by these advanced models guide experimental synthesis and testing with higher success rates, thereby accelerating the entire materials discovery process. Despite their power, DL methods often require larger datasets and can be less interpretable than traditional ML approaches. Ongoing research is focused on developing explainable DL models for materials science applications, aiming to combine the predictive power of deep learning with the interpretability needed for scientific insight.
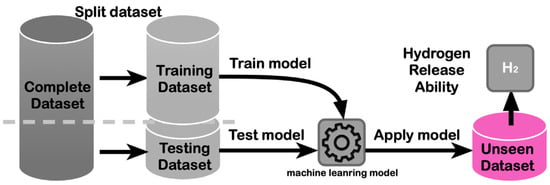
Figure 4.
Schematic representation of a machine learning (ML) workflow for predicting the hydrogen storage properties of materials. Reproduced with permission from [68], Elsevier, 2020.
In addition to the applications mentioned above, machine learning techniques are increasingly being utilized in two critical areas of hydrogen storage research: natural language processing (NLP) for information extraction. Natural language processing in hydrogen storage research leverages the vast amount of textual data available in scientific literature, patents, and technical reports. NLP techniques can automatically extract relevant information about material properties, synthesis conditions, and performance metrics from these sources. For instance, Krallinger et al. [69] describe how text mining and information retrieval technologies can be applied to chemical data, which are directly applicable to hydrogen storage materials. In our field, NLP can be used to automatically identify and extract information about new Mg-based alloys, their compositions, and reported hydrogen storage capacities from the published literature. This approach can significantly accelerate the discovery of promising new materials by efficiently processing large volumes of textual data.
By incorporating these advanced ML techniques, researchers can more efficiently navigate the vast chemical space of potential hydrogen storage materials, accelerate the materials discovery process, and optimize synthesis procedures. This integration of data-driven approaches with traditional experimental methods is paving the way for rapid advancements in the field of solid-state hydrogen storage.
Despite the promising applications of ML in hydrogen storage research, there are several challenges and limitations to be addressed. One of the main challenges is the availability and quality of training data. ML models rely on large and diverse datasets to learn meaningful patterns and make accurate predictions. However, the experimental and computational data on hydrogen storage materials is often limited, scattered, and inconsistent, making it difficult to compile comprehensive datasets for ML training. Efforts are being made to establish standardized databases and data sharing protocols to facilitate the development of ML models [70]. Another challenge is the interpretability of ML models. While ML models can make accurate predictions, they are often considered “black boxes” that provide little insight into the underlying physical mechanisms. Developing interpretable ML models that can unravel the structure-property relationships and provide explanations for their predictions is an active area of research [71]. Techniques such as feature importance analysis and visualization of learning patterns can help to improve the interpretability of ML models and guide the rational design of storage materials.
3. Assessment of Thermodynamic and Kinetic Properties
The thermodynamic and kinetic properties of hydrogen storage materials are key factors determining their practical applicability. The thermodynamic properties, such as the enthalpy and entropy of hydrogen absorption and desorption, dictate the temperature and pressure conditions required for hydrogen storage and release. The kinetic properties, such as the hydrogen absorption and desorption rates, determine the speed and reversibility of the storage process. Computational methods play a crucial role in assessing these properties and guiding the design of materials with optimal thermodynamic and kinetic characteristics.
DFT calculations are widely employed in the thermodynamic property assessment of hydrogen storage materials. By computing the total energy of hydrogenated and dehydrogenated phases, the energy changes during hydrogen absorption and desorption processes can be determined, thereby deriving the enthalpies of absorption and desorption [72]. These values are crucial for evaluating the stability and reversibility of materials during the storage process [73]. Additionally, statistical thermodynamics can be applied to estimate the entropy change during hydrogen storage by analyzing vibrational frequencies and configurational degrees of freedom. These thermodynamic quantities not only provide insights into the behavior of hydrogen storage materials under different operational conditions but also aid in identifying optimal hydrogen storage reaction conditions [74]. The optimal reaction enthalpy refers to the conditions (e.g., temperature, pressure) under which the hydrogen absorption and desorption processes of hydrogen storage materials are most effective and reliable. Thermodynamic quantities based on DFT calculations offer profound insights into the thermodynamic properties of hydrogen storage materials, providing essential theoretical foundations for the design and optimization of these materials. Based on DFT, Assila et al. [75] investigated the thermodynamic properties of Mg2Ni or Mg2NiH4 under various uniaxial and biaxial strain conditions. The results indicate that uniaxial/biaxial tensile and compressive strains induce structural deformations by increasing the strain thresholds of Mg2Ni and Mg2NiH4. Due to the contribution of strain energy, these strain effects on Mg2NiH4 can reduce the stability of the system more effectively than those on Mg2Ni, resulting in a decrease in formation enthalpy to −40 kJ/mol.
Table 5 presents a comparison of the calculated and experimentally measured enthalpies of hydrogen desorption for selected storage materials. The good agreement between the computational and experimental values demonstrates the capability of DFT calculations in predicting the thermodynamic properties of storage materials.
Table 5.
Comparison of calculated and experimentally measured enthalpies of hydrogen desorption for selected storage materials.
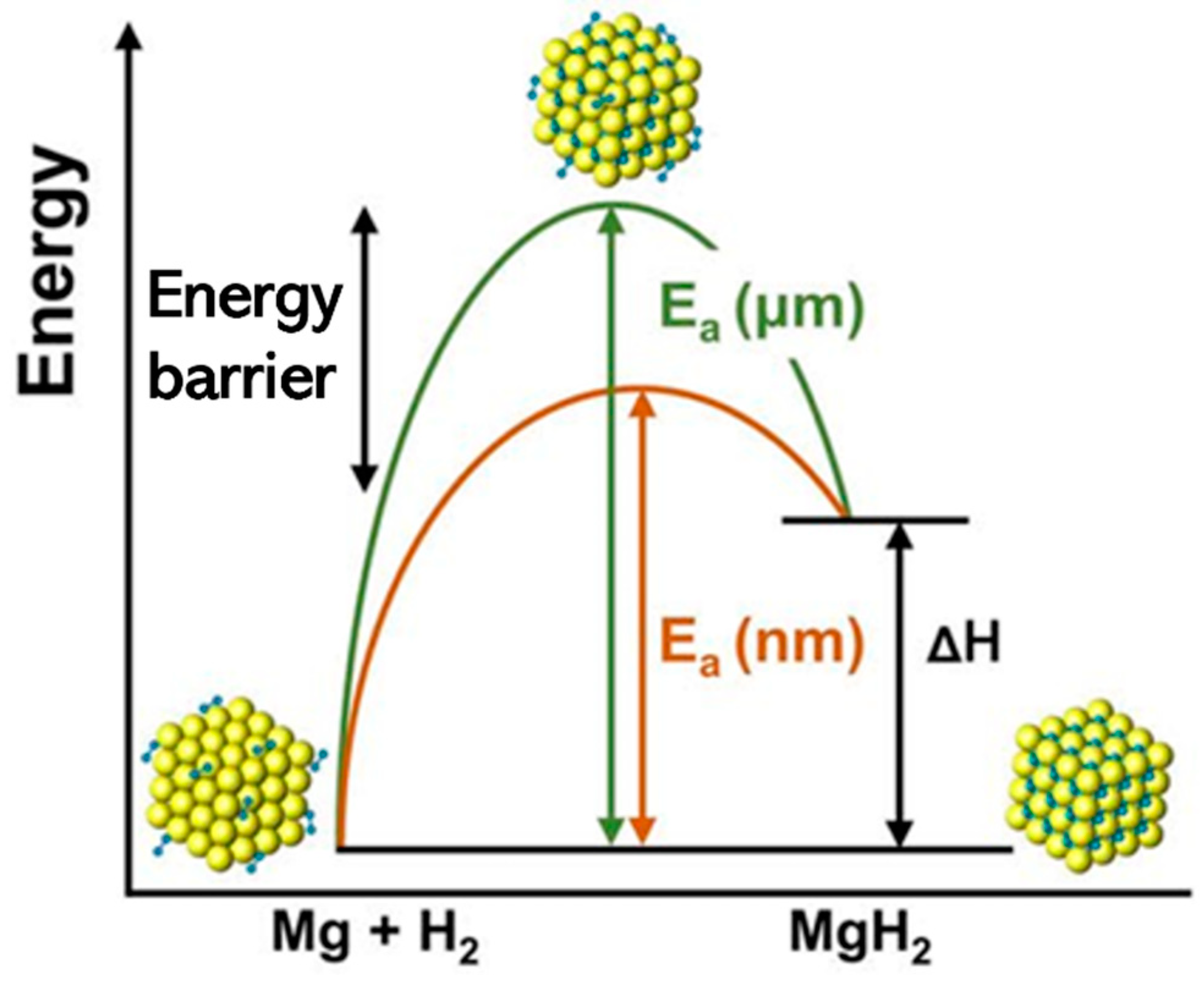
Kinetic properties, such as the hydrogen absorption and desorption rates, are often assessed using transition state theory (TST) and kinetic Monte Carlo (KMC) simulations. TST provides a framework for calculating the reaction rates based on the energy barriers and vibrational frequencies of the transition states. KMC simulations, on the other hand, can model the time evolution of the system by stochastically simulating the elementary reaction steps and diffusion events. By combining TST and KMC simulations with DFT calculations of the energy barriers, the kinetics of hydrogen storage can be predicted and optimized [76]. Figure 5 shows a schematic representation of the energy landscape for hydrogen absorption and desorption in a metal hydride. The energy barriers for the forward and reverse reactions determine the kinetics of the storage process. Computational methods can provide insights into the transition states and guide the design of materials with low energy barriers and fast kinetics [77].

Figure 5.
Schematic representation of the energy landscape for hydrogen absorption and desorption in a metal hydride. Reproduced with permission from [77], Springer Nature, 2023.
In addition to the assessment of the intrinsic thermodynamic and kinetic properties, computational methods can also investigate the influence of external factors, such as pressure, temperature, and catalysts, on the storage performance. For example, DFT calculations can elucidate the role of surface catalysts in lowering the energy barriers for hydrogen dissociation and recombination [78]. MD simulations can provide insights into the effect of temperature on hydrogen diffusion and desorption kinetics [79]. These computational studies guide the optimization of storage materials and the design of efficient storage systems.
4. Prediction of Stable Crystal Structures
The prediction of stable crystal structures is a crucial step in the discovery and design of novel hydrogen storage materials. The crystal structure determines the arrangement of atoms in the material and plays a significant role in the hydrogen storage properties, such as the binding energies, diffusion pathways, and thermodynamic stability. Computational methods, particularly those based on DFT, have become indispensable tools for predicting the stable crystal structures of storage materials.
One of the most widely used approaches for crystal structure prediction is the ab initio random structure searching (AIRSS) method [80]. AIRSS involves generating a large number of random initial structures and optimizing them using DFT calculations to find low-energy configurations. By exploring a diverse range of structural possibilities, AIRSS can identify the most thermodynamically stable crystal structures without prior knowledge of the material’s composition or symmetry. Another powerful approach for crystal structure prediction is the evolutionary algorithm (EA) [81]. EA mimics the process of natural evolution by generating a population of candidate structures and iteratively applying selection, mutation, and crossover operations to evolve towards the most stable configurations. EA has been successfully applied to predict the crystal structures of a wide range of materials, including hydrides and MOFs [82].
Moreover, classification methods play a crucial role in crystal structure prediction for hydrogen storage materials. Common approaches include support vector machines (SVMs), random forests (RFs), and gradient-boosting machines (GBMs). These methods utilize various descriptors to predict crystal structures, including: (1) Compositional descriptors: elemental properties, atomic radii, electronegativity; (2) structural descriptors: bond lengths, angles, coordination numbers; (3) electronic descriptors: band gap, density of states; (4) thermodynamic descriptors: formation energy, decomposition energy. However, it’s crucial to address the uncertainty in these predictions. Bayesian methods, such as Gaussian process classification, provide not just predictions but also confidence intervals, allowing researchers to quantify the reliability of crystal structure predictions. The statistical performance of these models is typically evaluated using metrics such as accuracy, precision, recall, and F1-score. For regression tasks, such as predicting lattice parameters, mean absolute error (MAE) and root mean square error (RMSE) are commonly used. It’s important to note that model performance can vary significantly depending on the dataset size, feature selection, and the specific materials system under study.
Table 6 presents a comparison of the predicted and experimentally observed crystal structures of selected hydrogen storage materials. The good agreement between the computational predictions and experimental observations demonstrates the capability of crystal structure prediction methods in identifying the stable phases of storage materials.
Table 6.
Comparison of predicted and experimentally observed crystal structures of selected hydrogen storage materials.
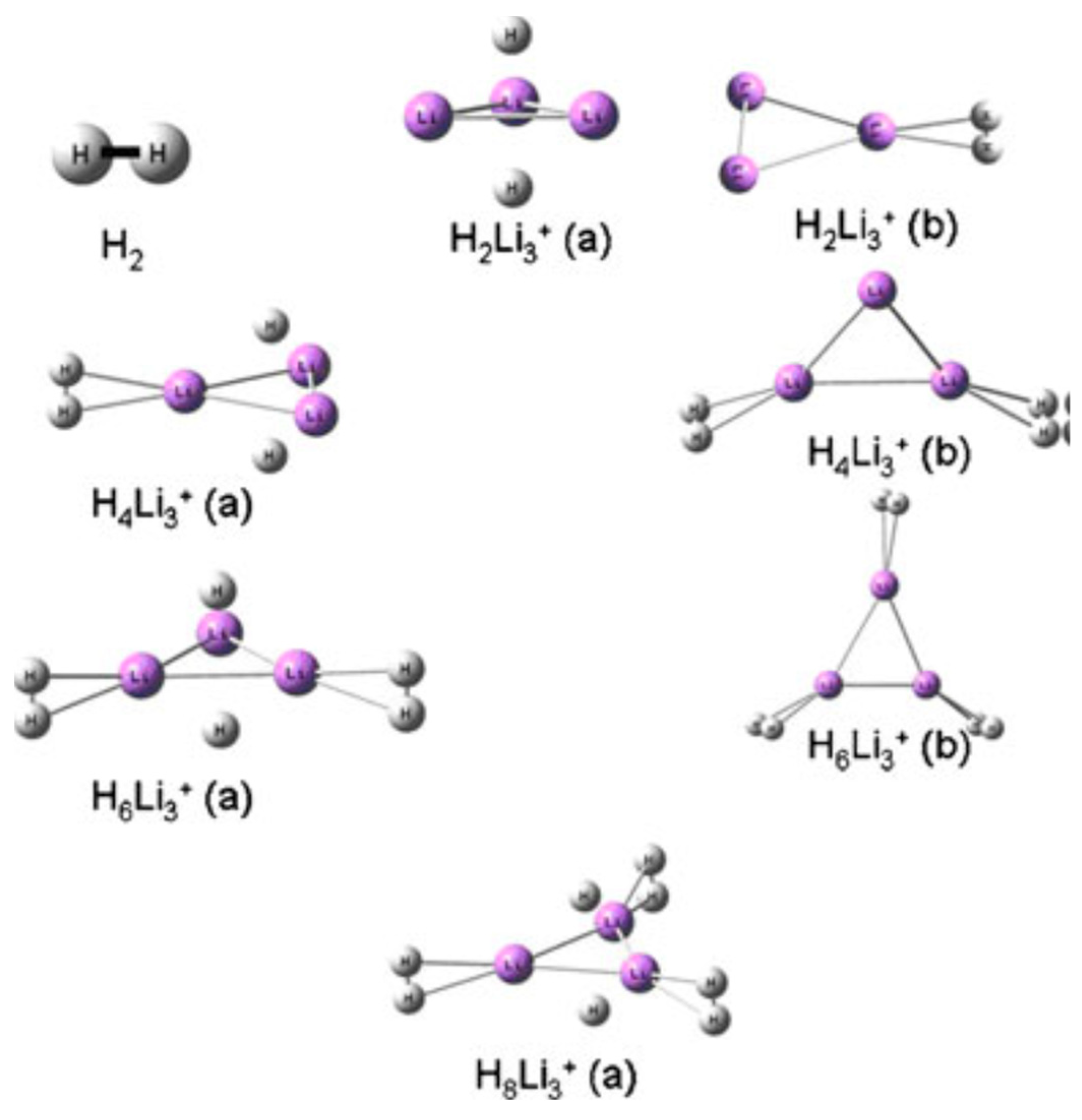
The prediction of stable crystal structures is particularly important for understanding phase transitions and structural changes during hydrogen absorption and desorption processes. Many hydrogen storage materials undergo significant structural rearrangements upon hydrogenation, often accompanied by changes in the crystal symmetry and lattice parameters. Computational methods can provide insights into these structural transformations and help identify the intermediate phases and transition pathways. Figure 6 illustrates the DFT-predicted crystal structures of various geometric conformations of the multi-hydrogen-bonded Li3+ complex. The figure also indicates that most H2 molecules captured within the triangular ring largely dissociate and ultimately become trapped in atomic form, whereas those bonded at the apex retain their diatomic nature, rather than exhibiting molecular properties [83].
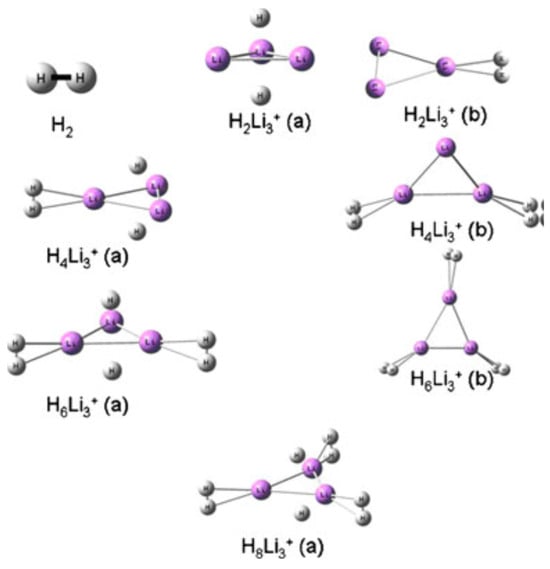
Figure 6.
Geometries of the different hydrogen trapped Li3+ (types a and b) clusters optimized at B3LYP/6–311+G(d) level of theory. Reproduced with permission from [83], Springer Nature, 2011.
In addition to predicting ground-state crystal structures, computational methods can also explore the metastable phases and polymorphs of storage materials. Metastable phases, although thermodynamically less stable than the ground state, can exhibit favorable kinetic properties or improved storage capacities [84]. By systematically searching the potential energy landscape, computational methods can identify metastable phases that may be overlooked by experiments and guide the synthesis of materials with tailored properties.
5. High-Throughput Screening and Materials Discovery
The discovery of novel hydrogen storage materials with improved performance is a critical challenge in the field. Traditional experimental approaches based on trial-and-error and intuition are often time-consuming and resource intensive. Computational methods, particularly high-throughput screening (HTS) techniques, have emerged as powerful tools to accelerate the materials discovery process [85].
HTS involves the automated and systematic screening of a large number of candidate materials using computational methods. By leveraging the efficiency and accuracy of DFT calculations, HTS can evaluate the hydrogen storage properties, such as the binding energies, thermodynamic stability, and storage capacities, of thousands of materials in a relatively short time [86]. The most promising candidates identified from the HTS can then be prioritized for experimental synthesis and testing, significantly reducing the experimental efforts and costs.
One of the key enablers of HTS is the development of materials databases and computational workflows. Materials databases, such as the Materials Project [87], NOMAD [88], and AiiDA [89], provide access to a vast amount of computational data on the structural, electronic, and thermodynamic properties of materials. These databases serve as a foundation for HTS by providing the necessary input data for the screening calculations. Computational workflows automate the various steps involved in the HTS process, from the generation of candidate structures to the analysis and visualization of the results [90].
HTS has led to the discovery of several promising hydrogen storage materials that were previously unknown or overlooked. Despite the success of HTS in accelerating materials discovery, there are several challenges and limitations to be addressed. One of the main challenges is the accuracy and reliability of the computational methods used in the screening process [91]. DFT calculations, although highly accurate for many materials, may not capture all the relevant physics, particularly for complex systems with strong correlation effects or weak interactions [92]. The development of more advanced electronic structure methods and the incorporation of machine learning techniques can help improve the accuracy and transferability of screening calculations [92].
Another challenge is the limited scope of the current HTS studies. Most HTS studies focus on a specific class of materials, such as MOFs or metal hydrides, and may overlook other promising material families. Expanding the material space and considering a broader range of materials, including novel and unconventional systems, can lead to the discovery of new storage materials with unique properties [93].
6. Multiscale Modeling and Experimental Validation
Hydrogen storage involves processes spanning multiple length and time scales, from atomic-level interactions to the macroscopic behavior of the storage system. To fully understand and optimize the performance of storage materials, it is essential to develop multiscale modeling frameworks that can bridge these scales and provide a comprehensive description of the storage processes.
Multiscale modeling approaches combine different computational methods, such as DFT calculations, MD simulations, and continuum-level models, to capture the relevant phenomena at each scale. At the atomic scale, DFT calculations provide insights into the electronic structure, binding energies, and reaction mechanisms. MD simulations can then capture the dynamics of hydrogen diffusion and sorption at the nanoscale. At the mesoscale, phase-field models and finite element methods can describe the microstructural evolution and mechanical behavior of the storage material [94]. Finally, at the macroscopic scale, continuum-level models, such as computational fluid dynamics (CFD) and heat transfer simulations, can predict the performance of the storage system under realistic operating conditions [95].
One of the key challenges in multiscale modeling is the transfer of information across different scales. The properties and behaviors at one scale often depend on the phenomena occurring at other scales. For example, the macroscopic hydrogen storage capacity is determined by the atomic-level interactions and the microstructural features of the material. Developing robust and efficient coupling schemes that can seamlessly
Another important aspect of multiscale modeling is the experimental validation of the computational predictions. Experimental techniques, such as X-ray diffraction, neutron scattering, and spectroscopic methods, provide valuable insights into the structure, dynamics, and thermodynamics of storage materials. By comparing the computational results with experimental data, the accuracy and reliability of the multiscale models can be assessed and improved. Experimental validation is not only crucial for assessing the accuracy of the computational models but also for guiding their development and refinement [96,97]. Discrepancies between the computational predictions and experimental observations can identify the limitations of the current models and highlight the areas that require further improvement. For example, if the computationally predicted hydrogen storage capacity differs significantly from the experimental measurements, it may indicate the need to include additional physical phenomena, such as surface effects or defects, in the computational models [98,99].
Multiscale modeling, when combined with experimental validation, can provide a powerful framework for understanding and optimizing hydrogen storage materials. By leveraging the strengths of different computational methods and experimental techniques, researchers can gain a comprehensive understanding of storage processes and develop strategies for designing high-performance materials.
7. Conclusions and Future Perspectives
This article delves into the key computational methods in solid-state hydrogen storage material research and their crucial role in understanding material performance and design. DFT, as a primary tool, accurately computes the electronic structure, band structure, and reactive sites of solid-state hydrogen storage materials, laying the foundation for theoretical predictions and material design. Despite its high computational costs in handling large systems and weak interactions, significant progress has been made through continuous optimization and approximation methods. MD simulations, by modeling atomic and molecular movements over time scales, reveal the structural dynamics and hydrogen adsorption behavior of solid-state hydrogen storage materials under varying temperatures and pressures. This approach not only validates theoretical model predictions but also provides dynamic information inaccessible through experiments, aiding in optimizing material thermodynamic and kinetic properties. ML techniques are increasingly applied in solid-state hydrogen material research to analyze large datasets and identify critical structure–property relationships, accelerating material screening and optimization processes. ML models handle complex nonlinear relationships, extracting hidden patterns and rules through extensive data training to enhance hydrogen absorption performance and stability, thereby saving considerable experimental and computational time. In summary, these computational methods not only deepen and broaden the understanding of solid-state hydrogen material properties but also provide multi-dimensional analysis tools for material design and development.
In future solid-state hydrogen material research, advancements will not be limited to the optimization of individual methods but will involve interdisciplinary collaboration to elevate overall research standards. In DFT, in addition to refining computational methods and enhancing accuracy, there is potential to explore the development of efficient energy functionals based on machine learning. This approach utilizes data-driven methods to rapidly optimize exchange-correlation energies, significantly improving computational speed and the reliability of simulation results. Concurrently, validating and refining DFT simulation results with experimental data ensures their accuracy and applicability in practical scenarios. In the field of molecular dynamics simulations, emphasis should be placed on the precision of force field parameters and model complexity. Quantum mechanical calculations can aid in optimizing force field parameters, better capturing material dynamic behaviors, and hydrogen adsorption/desorption processes. The application of parallel computing and optimization algorithms will further enhance MD simulation efficiency, enabling the handling of larger and more complex systems to provide more accurate dynamic information for material design. Regarding machine learning, besides enhancing dataset quality and diversity, exploring methods like incremental learning and adaptive model updates is crucial. Establishing dynamic data learning platforms can track real-time changes in data and knowledge, continuously optimizing models and improving prediction reliability. Moreover, leveraging the success of deep learning models such as convolutional neural networks (CNNs) and recurrent neural networks (RNNs) in image and sequence data processing can offer new insights and tools for predicting the structure and performance of solid-state hydrogen materials. Combining deep learning’s nonlinear processing capabilities with the stability of traditional physical models can achieve better results in complex structure analysis and feature extraction.
Furthermore, as artificial intelligence continues to evolve, embedding more advanced AI techniques in solid-state hydrogen material research holds tremendous potential. AI-driven approaches in material discovery, performance prediction and optimization, intelligent experiment design, material structure, and dynamic analysis, as well as interdisciplinary cooperation and knowledge integration, enable in-depth analysis and application. Future solutions could involve establishing an integrated AI platform focused on the design, simulation, and optimization of solid-state hydrogen materials. This platform would combine advanced machine learning algorithms with high-performance computing technology to rapidly identify and optimize potential high-efficiency hydrogen storage materials through big data analysis and experimental results. Multiscale simulation and prediction capabilities will deepen understanding of complex material structures and dynamic behaviors, providing precise guidance for material design. Real-time feedback and knowledge integration will promote interdisciplinary collaboration, address complex scientific challenges in solid-state hydrogen material research, and ultimately support the sustainable application and development of clean energy technologies.
Future advancements in solid-state hydrogen material research will heavily rely on technological integration and innovation, optimizing existing computational methods and introducing new technologies to achieve comprehensive understanding and precise predictions of material performance. The combination of DFT and MD will continue to enhance our understanding of material structure and dynamic behavior, while ML applications hold promise in accelerating material discovery and optimization processes, revealing hidden structure–property relationships through big data analysis. These advancements will not only facilitate the development of more efficient and stable hydrogen storage materials but also lay a solid foundation for the sustainable development of hydrogen energy technologies. Through precise predictions of key performance indicators such as hydrogen absorption capacity, stability, and cycle life, we can drive the commercialization process of new materials, thereby promoting widespread application of clean energy technologies. Additionally, strengthened interdisciplinary collaboration and integration of knowledge from different fields will enable comprehensive responses to global energy challenges. By fostering collaborative exchanges, experts from various fields can collectively address complex scientific problems, driving the convergence of materials science, chemical engineering, and computer science to provide critical support and innovative directions for the sustainable development of future energy.
Author Contributions
Conceptualization, X.Y. and H.J.; validation, Y.L. (Yuting Li); formal analysis, Y.L. (Yitao Liu), Q.L. and T.Y.; data curation, Y.L. (Yuting Li), T.Y. and Y.L. (Yitao Liu); writing—original draft preparation, X.Y., T.Y. and Y.L. (Yitao Liu); writing—review and editing, Q.L. and H.J.; supervision, Q.L., T.Y. and H.J.; project administration, X.Y. and H.J.; funding acquisition, X.Y. and H.J. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financially supported by Yunnan Province Major Science and Technology Project—Energy Conservation, Environmental Protection and New Energy (Project No.: 202402AF080001), Fundamental Research Funds for the Central Universities (2023CDJXY-019).
Data Availability Statement
Data available in a publicly accessible repository.
Conflicts of Interest
Authors Xi Yang and Tingna Yang were employed by the Yunnan Energy Research Institute Co., Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef]
- Ding, Z.; Lu, Y.; Li, L.; Shaw, L. High reversible capacity hydrogen storage through Nano-LiBH4 + Nano-MgH2 system. Energy Storage Mater. 2019, 20, 24–35. [Google Scholar] [CrossRef]
- Eberle, U.; Felderhoff, M.; Schüth, F. Chemical and Physical Solutions for Hydrogen Storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef]
- Ding, Z.; Shaw, L. Enhancement of Hydrogen Desorption from Nanocomposite Prepared by Ball Milling MgH2 with In Situ Aerosol Spraying LiBH4. ACS Sustain. Chem. Eng. 2019, 7, 15064–15072. [Google Scholar] [CrossRef]
- Allendorf, M.D.; Hulvey, Z.; Gennett, T.; Ahmed, A.; Autrey, T.; Camp, J.; Cho, E.S.; Furukawa, H.; Haranczyk, M.; Head-Gordon, M.; et al. An assessment of strategies for the development of solid-state adsorbents for vehicular hydrogen storage. Energy Environ. Sci. 2018, 11, 2784–2812. [Google Scholar] [CrossRef]
- Massaro, M.C.; Biga, R.; Kolisnichenko, A.; Marocco, P.; Monteverde, A.H.A.; Santarelli, M. Potential and technical challenges of on-board hydrogen storage technologies coupled with fuel cell systems for aircraft electrification. J. Power Sources 2023, 555, 232397. [Google Scholar] [CrossRef]
- Ding, Z.; Li, H.; Shaw, L. New insights into the solid-state hydrogen storage of nanostructured LiBH4-MgH2 system. Chem. Eng. J. 2020, 385, 123856. [Google Scholar] [CrossRef]
- Jhi, S.-H.; Kwon, Y.-K.; Bradley, K.; Gabriel, J.-C.P. Hydrogen storage by physisorption: Beyond carbon. Solid State Commun. 2004, 129, 769–773. [Google Scholar] [CrossRef]
- Kumar, A.; Muthukumar, P.; Sharma, P.; Kumar, E.A. Absorption based solid state hydrogen storage system: A review. Sustain. Energy Technol. Assess. 2022, 52, 102204. [Google Scholar] [CrossRef]
- Liu, S.; Liu, J.; Liu, X.; Shang, J.-X.; Yu, R.; Shui, J. Non-classical hydrogen storage mechanisms other than chemisorption and physisorption. Appl. Phys. Rev. 2022, 9, 021315. [Google Scholar] [CrossRef]
- Chen, Z.; Kirlikovali, K.O.; Idrees, K.B.; Wasson, M.C.; Farha, O.K. Porous materials for hydrogen storage. Chem 2022, 8, 693–716. [Google Scholar] [CrossRef]
- Zacharia, R.; Rather, S.U. Review of Solid State Hydrogen Storage Methods Adopting Different Kinds of Novel Materials. J. Nanomater. 2015, 2015, 914845. [Google Scholar] [CrossRef]
- Ding, Z.; Li, Y.; Yang, H.; Lu, Y.; Tan, J.; Li, J.; Li, Q.; Chen, Y.A.; Shaw, L.L.; Pan, F. Tailoring MgH2 for hydrogen storage through nanoengineering and catalysis. J. Magnes. Alloys 2022, 10, 2946–2967. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, C.G.; Zhou, H.; Ye, E.; Xu, J.; Li, Z.; Loh, X.J. Current Research Trends and Perspectives on Solid-State Nanomaterials in Hydrogen Storage. Research 2021, 2021, 3750689. [Google Scholar] [CrossRef]
- Singh, S.B.; De, M. Alumina based doped templated carbons: A comparative study with zeolite and silica gel templates. Microporous Mesoporous Mater. 2018, 257, 241–252. [Google Scholar] [CrossRef]
- Ahmed, A.; Seth, S.; Purewal, J.; Wong-Foy, A.G.; Veenstra, M.; Matzger, A.J.; Siegel, D.J. Exceptional hydrogen storage achieved by screening nearly half a million metal-organic frameworks. Nat. Commun. 2019, 10, 1568. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Q.; Ding, Z.; Jiang, H.; Yang, H.; Du, W.; Zheng, Y.; Huo, K.; Shaw, L.L. MOFs-Based Materials for Solid-State Hydrogen Storage: Strategies and Perspectives. Chem. Eng. J. 2024, 485, 149665. [Google Scholar] [CrossRef]
- Samantaray, S.S.; Putnam, S.T.; Stadie, N.P. Volumetrics of Hydrogen Storage by Physical Adsorption. Inorganics 2021, 9, 45. [Google Scholar] [CrossRef]
- Ding, Z.; Li, S.; Zhou, Y.; Chen, Z.; Yang, W.; Ma, W.; Shaw, L. LiBH4 for hydrogen storage—New perspectives. Nano Mater. Sci. 2020, 2, 109–119. [Google Scholar] [CrossRef]
- Singh, S.B.; De, M. Thermally exfoliated graphene oxide for hydrogen storage. Mater. Chem. Phys. 2020, 239, 122102. [Google Scholar] [CrossRef]
- Singh, S.B.; De, M. Effects of gaseous environments on physicochemical properties of thermally exfoliated graphene oxides for hydrogen storage: A comparative study. J. Porous Mater. 2021, 28, 875–888. [Google Scholar] [CrossRef]
- Majid, N.A.A.; Watanabe, J.; Notomi, M. Improved desorption temperature of magnesium hydride via multi-layering Mg/Fe thin film. Int. J. Hydrogen Energy 2021, 46, 4181–4187. [Google Scholar] [CrossRef]
- Baum, L.; Meyer, M.; Mendoza-Zélis, L. Hydrogen storage properties of the Mg/Fe system. Phys. B Condens. Matter 2007, 389, 189–192. [Google Scholar] [CrossRef]
- Reardon, H.; Hanlon, J.M.; Hughes, R.W.; Godula-Jopek, A.; Mandal, T.K.; Gregory, D.H. Emerging concepts in solid-state hydrogen storage: The role of nanomaterials design. Energy Environ. Sci. 2012, 5, 5951–5979. [Google Scholar] [CrossRef]
- Rönnebro, E.C.E.; Majzoub, E.H. Recent advances in metal hydrides for clean energy applications. MRS Bull. 2013, 38, 452–458. [Google Scholar] [CrossRef]
- Jordá-Beneyto, M.; Suárez-García, F.; Lozano-Castelló, D.; Cazorla-Amorós, D.; Linares-Solano, A. Hydrogen storage on chemically activated carbons and carbon nanomaterials at high pressures. Carbon 2007, 45, 293–303. [Google Scholar] [CrossRef]
- Kishor, R.; Singh, S.B.; Ghoshal, A.K. Role of metal type on mesoporous KIT-6 for hydrogen storage. Int. J. Hydrogen Energy 2018, 43, 10376–10385. [Google Scholar] [CrossRef]
- Balaprakash, P.; Dongarra, J.; Gamblin, T.; Hall, M.; Hollingsworth, J.K.; Norris, B.; Vuduc, R. Autotuning in High-Performance Computing Applications. Proc. IEEE 2018, 106, 2068–2083. [Google Scholar] [CrossRef]
- Correa-Baena, J.-P.; Hippalgaonkar, K.; van Duren, J.; Jaffer, S.; Chandrasekhar, V.R.; Stevanovic, V.; Wadia, C.; Guha, S.; Buonassisi, T. Accelerating Materials Development via Automation, Machine Learning, and High-Performance Computing. Joule 2018, 2, 1410–1420. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Dubecký, M.; Mitas, L.; Jurečka, P. Noncovalent Interactions by Quantum Monte Carlo. Chem. Rev. 2016, 116, 5188–5215. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Li, W.; Jiang, T.; Wang, Y.; Shuai, Z. Time-dependent density matrix renormalization group method for quantum dynamics in complex systems. WIREs Comput. Mol. Sci. 2022, 12, e1614. [Google Scholar] [CrossRef]
- Gao, Y.; Li, Z.; Wang, P.; Li, C.; Yue, Q.; Cui, W.G.; Wang, X.; Yang, Y.; Gao, F.; Zhang, M.; et al. Solid-State Hydrogen Storage Origin and Design Principles of Carbon-Based Light Metal Single-Atom Materials. Adv. Funct. Mater. 2024, 34, 2316368. [Google Scholar] [CrossRef]
- Psofogiannakis, G.M.; Froudakis, G.E. DFT Study of Hydrogen Storage by Spillover on Graphite with Oxygen Surface Groups. J. Am. Chem. Soc. 2009, 131, 15133–15135. [Google Scholar] [CrossRef]
- Marian, C.M.; Heil, A.; Kleinschmidt, M. The DFT/MRCI method. WIREs Comput. Mol. Sci. 2019, 9, e1394. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Feynman, R.P. Statistical Mechanics: A Set of Lectures; Benjamin/Cummings Publishing Company: Reading, MA, USA, 1972. [Google Scholar]
- Zhao, Y.; Zhu, Y.; Shi, R.; Jia, Z.; Zhang, J.; Liu, Y.; Cheng, H.; Tang, Q.; Ba, Z.; Hu, X.; et al. Structural inhomogeneity: A potential strategy to improve the hydrogen storage performance of metal hydrides. J. Mater. Chem. A 2023, 11, 13255–13265. [Google Scholar] [CrossRef]
- Ding, Z.; Li, H.; Yan, G.; Yang, W.; Gao, Z.; Ma, W.; Shaw, L. Mechanism of hydrogen storage on Fe3B. Chem. Commun. 2020, 56, 14235–14238. [Google Scholar] [CrossRef]
- Xue, W.; Zhao, B.; Liu, H.; Chen, X.; Liu, L. Ultralow Pd bimetallic catalysts boost (de)hydrogenation for reversible H2 storage. Appl. Catal. B Environ. 2024, 343, 123574. [Google Scholar] [CrossRef]
- Verma, P.; Truhlar, D.G. Status and Challenges of Density Functional Theory. Trends Chem. 2020, 2, 302–318. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef]
- Han, Z.; Wu, Y.; Yu, H.; Zhou, S. Location-dependent effect of nickel on hydrogen dissociation and diffusion on Mg (0001) surface: Insights into hydrogen storage material design. J. Magnes. Alloys 2022, 10, 1617–1630. [Google Scholar] [CrossRef]
- Zhu, B.-C.; Liu, G.-H.; Deng, P.-J.; Liu, C.-J.; Liao, Y.-H.; Zeng, L.; Zhao, J. Study of the hydrogen absorption behaviour of a “number-sensitive” Mg atom: Ultra-high hydrogen storage in MgHn (n = 1–20) clusters. J. Mater. Chem. A 2023, 11, 13774–13782. [Google Scholar] [CrossRef]
- Prasetyo, N.; Pambudi, F.I. Toward hydrogen storage material in fluorinated zirconium metal-organic framework (MOF-801): A periodic density functional theory (DFT) study of fluorination and adsorption. Int. J. Hydrogen Energy 2021, 46, 4222–4228. [Google Scholar] [CrossRef]
- Xia, L.; Bo, Z.; Liu, Q.; Zhang, X.; Pei, Y. Li-doped and functionalized metal-organic framework-519 for enhancing hydrogen storage: A computational study. Comput. Mater. Sci. 2019, 166, 179–186. [Google Scholar] [CrossRef]
- Kurth, S.; Perdew, J.P.; Blaha, P. Molecular and solid-state tests of density functional approximations: LSD, GGAs, and meta-GGAs. Int. J. Quantum Chem. 1999, 75, 889–909. [Google Scholar] [CrossRef]
- Klimeš, J.; Michaelides, A. Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory. J. Chem. Phys. 2012, 137, 120901. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Bowler, D.R.; Miyazaki, T. methods in electronic structure calculations. Rep. Prog. Phys. 2012, 75, 036503. [Google Scholar] [CrossRef]
- Cerutti, D.S.; Case, D.A. Molecular dynamics simulations of macromolecular crystals. WIREs Comput. Mol. Sci. 2019, 9, e1402. [Google Scholar] [CrossRef]
- Shi, R.; Qian, H.-J.; Lu, Z.-Y. Coarse-grained molecular dynamics simulation of polymers: Structures and dynamics. WIREs Comput. Mol. Sci. 2023, 13, e1683. [Google Scholar] [CrossRef]
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A., III; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Bai, S.; Piri, M. Hydrogen storage in nanoporous media: Molecular dynamics simulations of the confinement effects. Int. J. Hydrogen Energy 2022, 47, 24886–24896. [Google Scholar] [CrossRef]
- Akbarzadeh, F.Z.; Rajabi, M. Mechanical alloying fabrication of nickel/cerium/MgH2 nanocomposite for hydrogen storage: Molecular dynamics study and experimental verification. J. Alloys Compd. 2022, 899, 163280. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, Y.; Wang, X.-D.; Zhang, W.; Ma, E.; Deringer, V.L.; Mazzarello, R. Unraveling Crystallization Mechanisms and Electronic Structure of Phase-Change Materials by Large-Scale Ab Initio Simulations. Adv. Mater. 2022, 34, 2109139. [Google Scholar] [CrossRef]
- Reilly, J.J., Jr.; Wiswall, R.H., Jr. Reaction of hydrogen with alloys of magnesium and nickel and the formation of Mg2NiH4. Inorg. Chem. 1968, 7, 2254–2256. [Google Scholar] [CrossRef]
- Yartys, V.A.; Lototskyy, M.V.; Akiba, E.; Albert, R.; Antonov, V.E.; Ares, J.R.; Baricco, M.; Bourgeois, N.; Buckley, C.E.; von Colbe, J.M.B.; et al. Magnesium based materials for hydrogen based energy storage: Past, present and future. Int. J. Hydrogen Energy 2019, 44, 7809–7859. [Google Scholar] [CrossRef]
- Azizzadenesheli, K.; Kovachki, N.; Li, Z.; Liu-Schiaffini, M.; Kossaifi, J.; Anandkumar, A. Neural operators for accelerating scientific simulations and design. Nat. Rev. Phys. 2024, 6, 320–328. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Long time scale kinetic Monte Carlo simulations without lattice approximation and predefined event table. J. Chem. Phys. 2001, 115, 9657–9666. [Google Scholar] [CrossRef]
- Biamonte, J.; Wittek, P.; Pancotti, N.; Rebentrost, P.; Wiebe, N.; Lloyd, S. Quantum machine learning. Nature 2017, 549, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Salehi, K.; Rahmani, M.; Atashrouz, S. Machine learning assisted predictions for hydrogen storage in metal-organic frameworks. Int. J. Hydrogen Energy 2023, 48, 33260–33275. [Google Scholar] [CrossRef]
- Zhou, P.; Xiao, X.; Zhu, X.; Chen, Y.; Lu, W.; Piao, M.; Cao, Z.; Lu, M.; Fang, F.; Li, Z.; et al. Machine learning enabled customization of performance-oriented hydrogen storage materials for fuel cell systems. Energy Storage Mater. 2023, 63, 102964. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Xiang, X.; Zu, X.; Hu, S. Hydrogen diffusion in zirconium hydrides from on-the-fly machine learning molecular dynamics. Int. J. Hydrogen Energy 2024, 56, 1057–1066. [Google Scholar] [CrossRef]
- Ding, Z.; Chen, Z.; Ma, T.; Lu, C.-T.; Ma, W.; Shaw, L. Predicting the hydrogen release ability of LiBH4-based mixtures by ensemble machine learning. Energy Storage Mater. 2020, 27, 466–477. [Google Scholar] [CrossRef]
- Krallinger, M.; Rabal, O.; Lourenco, A.; Oyarzabal, J.; Valencia, A. Information Retrieval and Text Mining Technologies for Chemistry. Chem. Rev. 2017, 117, 7673–7761. [Google Scholar] [CrossRef]
- Dong, S.; Wang, Y.; Li, J.; Li, Y.; Wang, L.; Zhang, J. Exploration and design of Mg alloys for hydrogen storage with supervised machine learning. Int. J. Hydrogen Energy 2023, 48, 38412–38424. [Google Scholar] [CrossRef]
- Shah, S.S.A.; Zafar, H.K.; Javed, M.S.; Din, M.A.U.; Alarfaji, S.S.; Balkourani, G.; Sohail, M.; Tsiakaras, P.; Najam, T. Mxenes for Zn-based energy storage devices: Nano-engineering and machine learning. Coord. Chem. Rev. 2024, 501, 215565. [Google Scholar] [CrossRef]
- Hu, J.; Shen, H.; Jiang, M.; Gong, H.; Xiao, H.; Liu, Z.; Sun, G.; Zu, X. A DFT Study of Hydrogen Storage in High-Entropy Alloy TiZrHfScMo. Nanomaterials 2019, 9, 461. [Google Scholar] [CrossRef]
- Yang, J.; Sudik, A.; Wolverton, C.; Siegel, D.J. High capacity hydrogen storage materials: Attributes for automotive applications and techniques for materials discovery. Chem. Soc. Rev. 2010, 39, 656–675. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Baron, G.V.; Perreault, P.; Lenaerts, S.; Ciocarlan, R.-G.; Cool, P.; Mileo, P.G.M.; Rogge, S.; Van Speybroeck, V.; Watson, G.; et al. Hydrogen Clathrates: Next Generation Hydrogen Storage Materials. Energy Storage Mater. 2021, 41, 69–107. [Google Scholar] [CrossRef]
- Assila, A.; Rkhis, M.; Alaoui-Belghiti, A.; Laasri, S.; Hlil, E.K.; Boughaleb, Y.; Hajjaji, A. Feeling the strain: Enhancing the thermodynamics characteristics of magnesium nickel hydride Mg2NiH4 for hydrogen storage applications through strain engineering. Int. J. Hydrogen Energy 2024, 67, 651–657. [Google Scholar] [CrossRef]
- Andreasen, A.; Vegge, T.; Pedersen, A.S. Dehydrogenation kinetics of as-received and ball-milled LiAlH4. J. Solid State Chem. 2005, 178, 3672–3678. [Google Scholar] [CrossRef]
- Ren, L.; Li, Y.; Zhang, N.; Li, Z.; Lin, X.; Zhu, W.; Lu, C.; Ding, W.; Zou, J. Nanostructuring of Mg-Based Hydrogen Storage Materials: Recent Advances for Promoting Key Applications. Nano-Micro Lett. 2023, 15, 93. [Google Scholar] [CrossRef]
- Li, T.; Kim, M.; Liang, Z.; Asthagiri, A.; Weaver, J.F. Dissociative Chemisorption and Oxidation of H2 on the Stoichiometric IrO2(110) Surface. Top. Catal. 2018, 61, 397–411. [Google Scholar] [CrossRef]
- Bogdanović, B.; Schwickardi, M. Ti-doped alkali metal aluminium hydrides as potential novel reversible hydrogen storage materials. Invited paper presented at the International Symposium on Metal–Hydrogen Systems, Les Diablerets, August 25–30, 1996, Switzerland. J. Alloys Compd. 1997, 253–254, 1–9. [Google Scholar] [CrossRef]
- Pickard, C.J.; Needs, R.J. Ab initio random structure searching. J. Phys. Condens. Matter 2011, 23, 053201. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.W.; Oganov, A.R.; Hansen, N. USPEX—Evolutionary crystal structure prediction. Comput. Phys. Commun. 2006, 175, 713–720. [Google Scholar] [CrossRef]
- Lee, S.; Kim, B.; Cho, H.; Lee, H.; Lee, S.Y.; Cho, E.S.; Kim, J. Computational Screening of Trillions of Metal–Organic Frameworks for High-Performance Methane Storage. ACS Appl. Mater. Interfaces 2021, 13, 23647–23654. [Google Scholar] [CrossRef]
- Giri, S.; Chakraborty, A.; Chattaraj, P.K. Potential use of some metal clusters as hydrogen storage materials—A conceptual DFT approach. J. Mol. Model. 2011, 17, 777–784. [Google Scholar] [CrossRef]
- Li, J.; Li, B.; Yu, X.; Zhao, H.; Shao, H. Geometrical effect in Mg-based metastable nano alloys with BCC structure for hydrogen storage. Int. J. Hydrogen Energy 2019, 44, 29291–29296. [Google Scholar] [CrossRef]
- Curtarolo, S.; Hart, G.L.W.; Nardelli, M.B.; Mingo, N.; Sanvito, S.; Levy, O. The high-throughput highway to computational materials design. Nat. Mater. 2013, 12, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Curtarolo, S.; Setyawan, W.; Hart, G.L.W.; Jahnatek, M.; Chepulskii, R.V.; Taylor, R.H.; Wang, S.; Xue, J.; Yang, K.; Levy, O.; et al. AFLOW: An automatic framework for high-throughput materials discovery. Comput. Mater. Sci. 2012, 58, 218–226. [Google Scholar] [CrossRef]
- Spotte-Smith, E.W.C.; Cohen, O.A.; Blau, S.M.; Munro, J.M.; Yang, R.; Guha, R.D.; Patel, H.D.; Vijay, S.; Huck, P.; Kingsbury, R.; et al. A database of molecular properties integrated in the Materials Project. Digit. Discov. 2023, 2, 1862–1882. [Google Scholar] [CrossRef]
- Sbailò, L.; Fekete, Á.; Ghiringhelli, L.M.; Scheffler, M. The NOMAD Artificial-Intelligence Toolkit: Turning materials-science data into knowledge and understanding. npj Comput. Mater. 2022, 8, 250. [Google Scholar] [CrossRef]
- Muy, S.; Johnston, C.; Marzari, N. AiiDA-defects: An automated and fully reproducible workflow for the complete characterization of defect chemistry in functional materials. Electron. Struct. 2023, 5, 024009. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Li, X.-B.; Zheng, H.; Chen, N.-K.; Wang, X.-P.; Zhang, X.-L.; Sun, H.-B.; Zhang, S. High-Throughput Screening for Phase-Change Memory Materials. Adv. Funct. Mater. 2021, 31, 2009803. [Google Scholar] [CrossRef]
- Rodríguez-Martínez, X.; Pascual-San-José, E.; Campoy-Quiles, M. Accelerating organic solar cell material’s discovery: High-throughput screening and big data. Energy Environ. Sci. 2021, 14, 3301–3322. [Google Scholar] [CrossRef]
- Paul, A.; Birol, T. Applications of DFT + DMFT in Materials Science. Annu. Rev. Mater. Res. 2019, 49, 31–52. [Google Scholar] [CrossRef]
- Zheng, S.; Ding, H.; Li, S.; Chen, D.; Pan, F. Application of topology-based structure features for machine learning in materials science. Chin. J. Struct. Chem. 2023, 42, 100120. [Google Scholar] [CrossRef]
- Xie, C.; He, X.K.; Liu, X.; Ye, J.H.; Chen, J.B. Phase-field modeling for anisotropic ductile damage of magnesium alloys at finite deformations. J. Magnes. Alloys 2022, in press. [Google Scholar] [CrossRef]
- Yu, C.; Qin, S.; Chai, B.; Huang, S.; Liu, Y. The Effect of Compressible Flow on Heat Transfer Performance of Heat Exchanger by Computational Fluid Dynamics (CFD) Simulation. Entropy 2019, 21, 829. [Google Scholar] [CrossRef]
- Chen, C.; Nguyen, D.T.; Lee, S.J.; Baker, N.A.; Karakoti, A.S.; Lauw, L.; Owen, C.; Mueller, K.T.; Bilodeau, B.A.; Murugesan, V.; et al. Accelerating Computational Materials Discovery with Machine Learning and Cloud High-Performance Computing: From Large-Scale Screening to Experimental Validation. J. Am. Chem. Soc. 2024. [Google Scholar] [CrossRef]
- Waser, R.; Ghani, F.; Maranda, S.; O’Donovan, T.S.; Schuetz, P.; Zaglio, M.; Worlitschek, J. Fast and experimentally validated model of a latent thermal energy storage device for system level simulations. Appl. Energy 2018, 231, 116–126. [Google Scholar] [CrossRef]
- Altintas, C.; Keskin, S. On the shoulders of high-throughput computational screening and machine learning: Design and discovery of MOFs for H2 storage and purification. Mater. Today Energy 2023, 38, 101426. [Google Scholar] [CrossRef]
- Basdogan, Y.; Keskin, S. Simulation and modelling of MOFs for hydrogen storage. CrystEngComm 2015, 17, 261–275. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).