Plasma Exfoliated Graphene: Preparation via Rapid, Mild Thermal Reduction of Graphene Oxide and Application in Lithium Batteries



{kind=link}
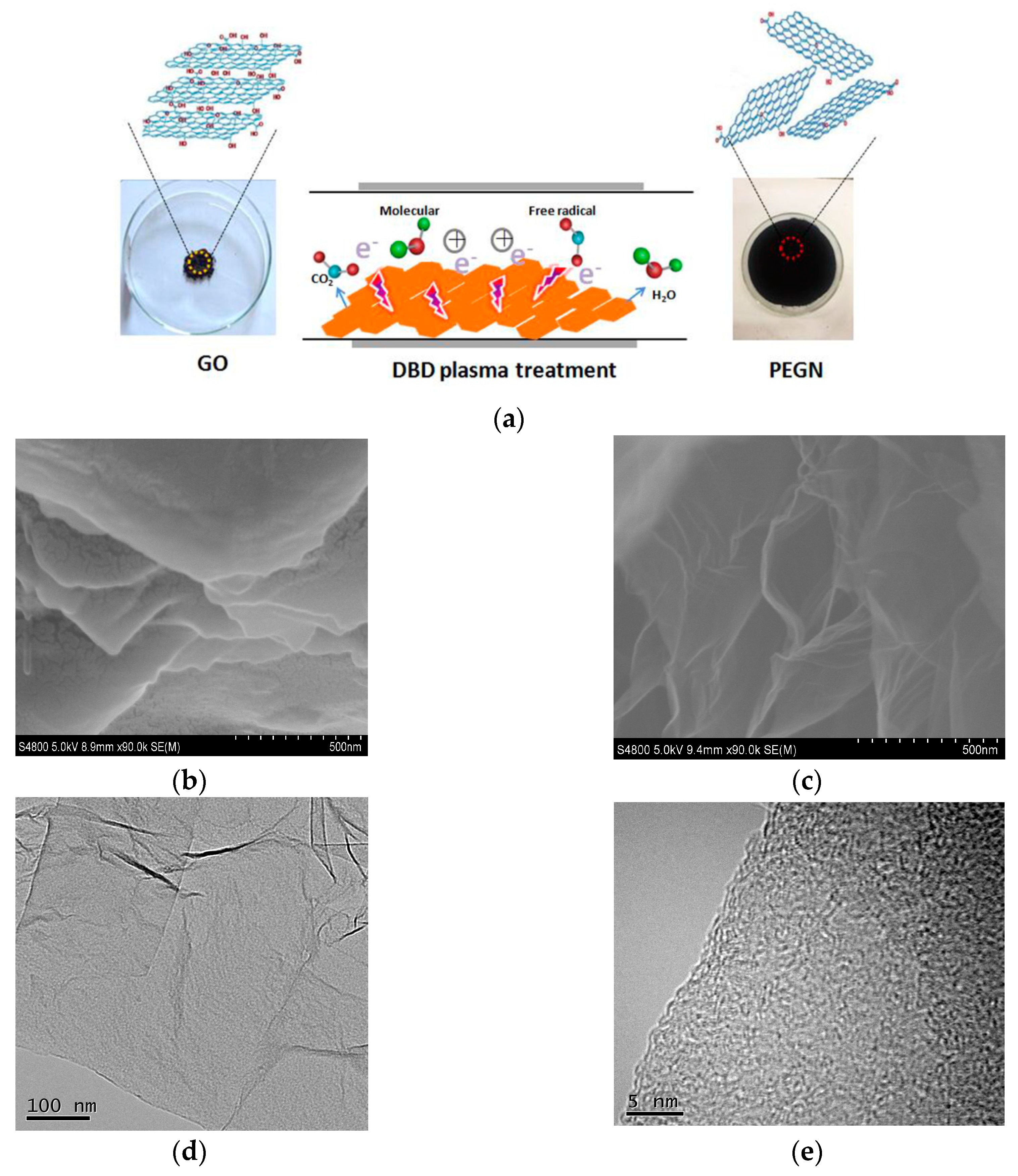{kind=link}
{kind=link}
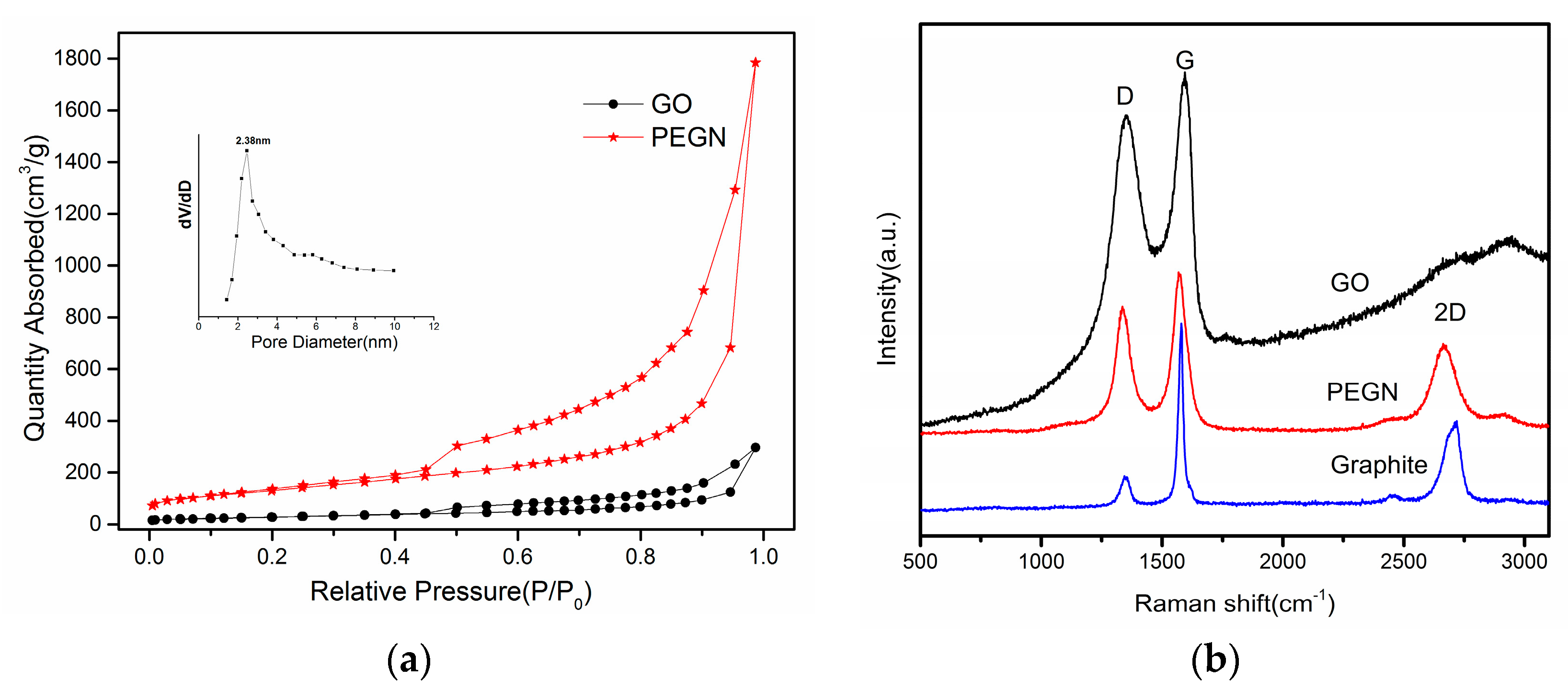{kind=link}
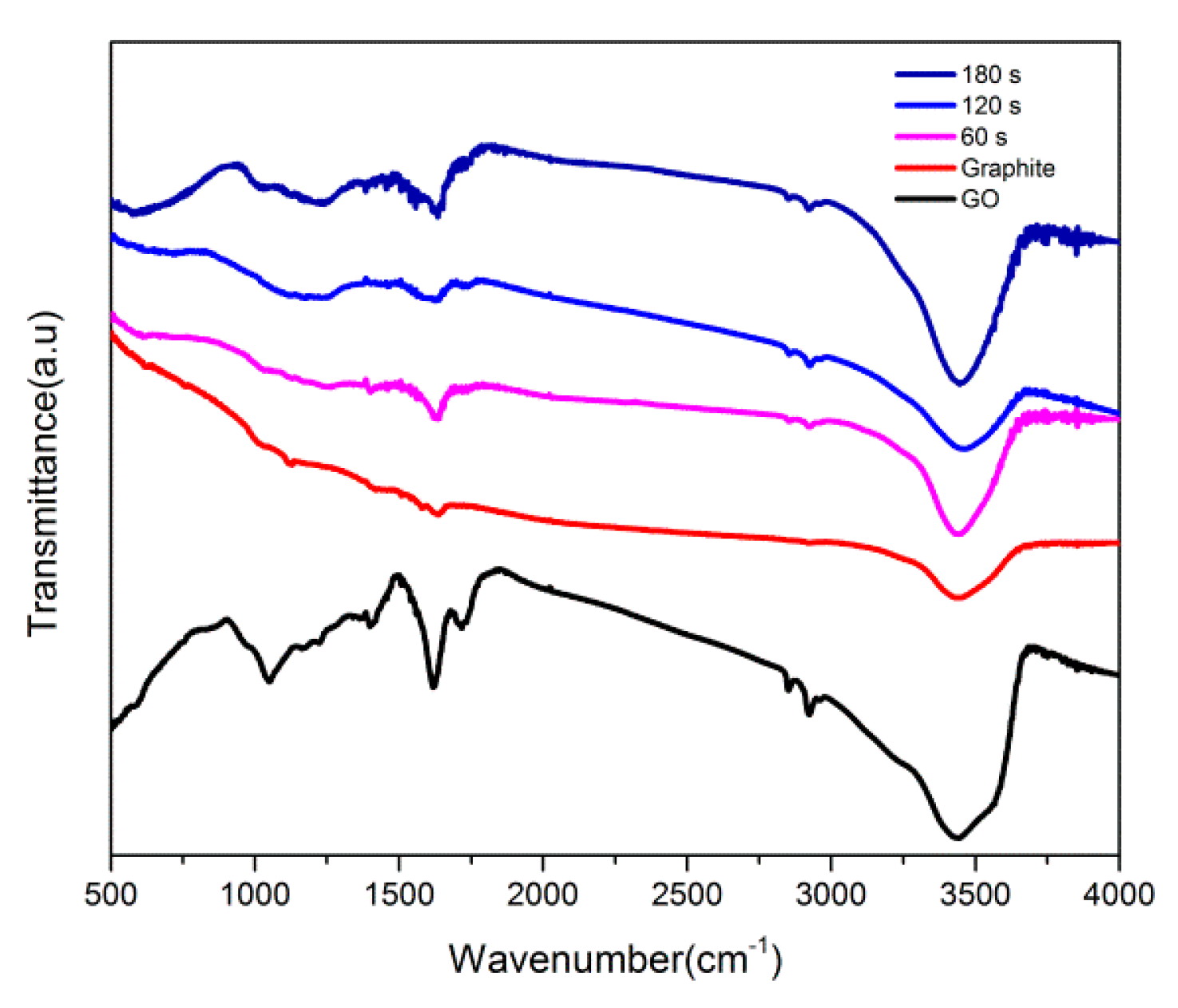{kind=link}
{kind=link}
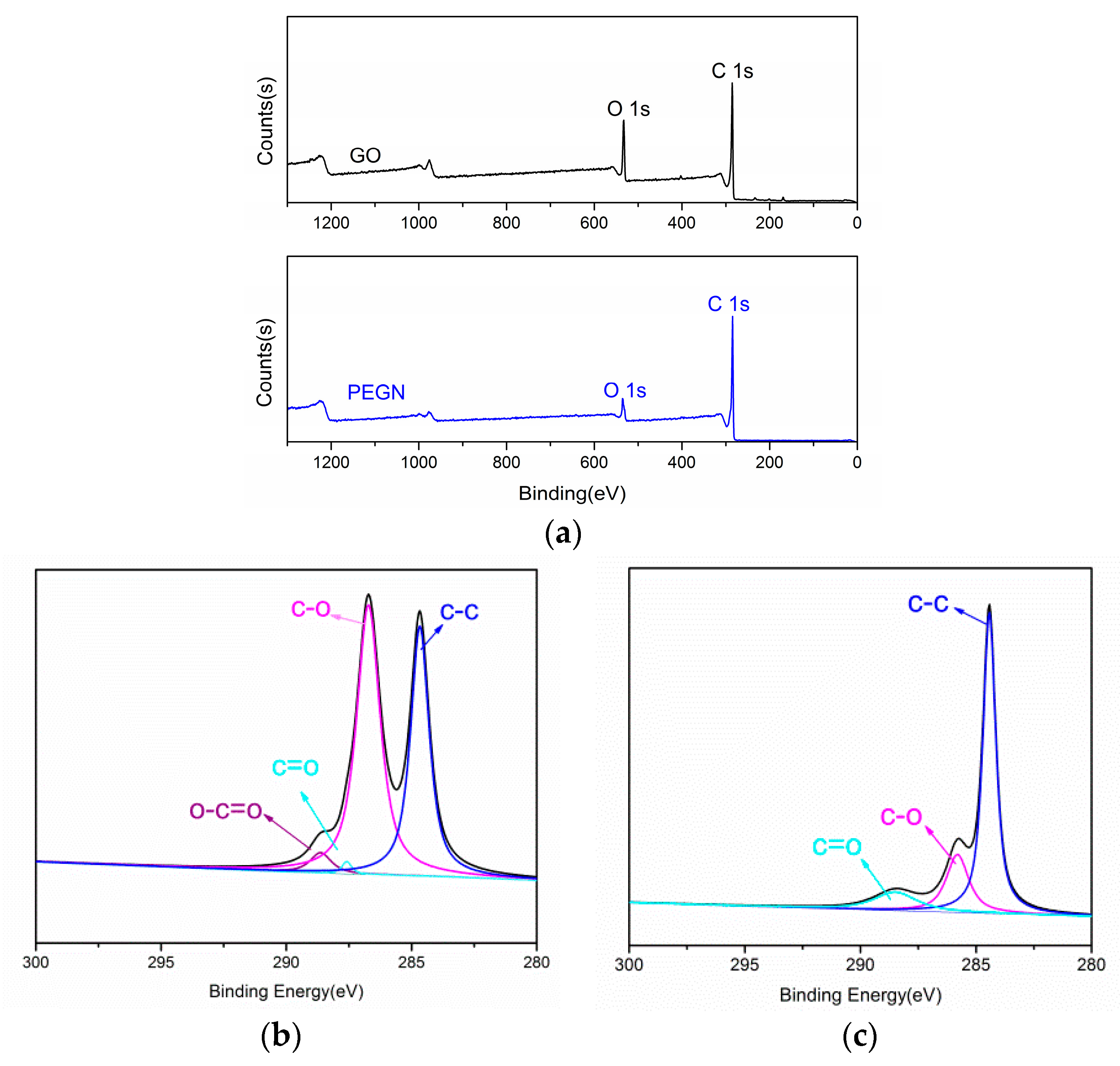{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Preparation of GO
2.3. Preparation of the PEGN with DBD Plasma
2.4. Characterization of PEGN
2.5. Electrochemical Measurement of PEGN
3. Results and Discussion
3.1. Characterization Studies
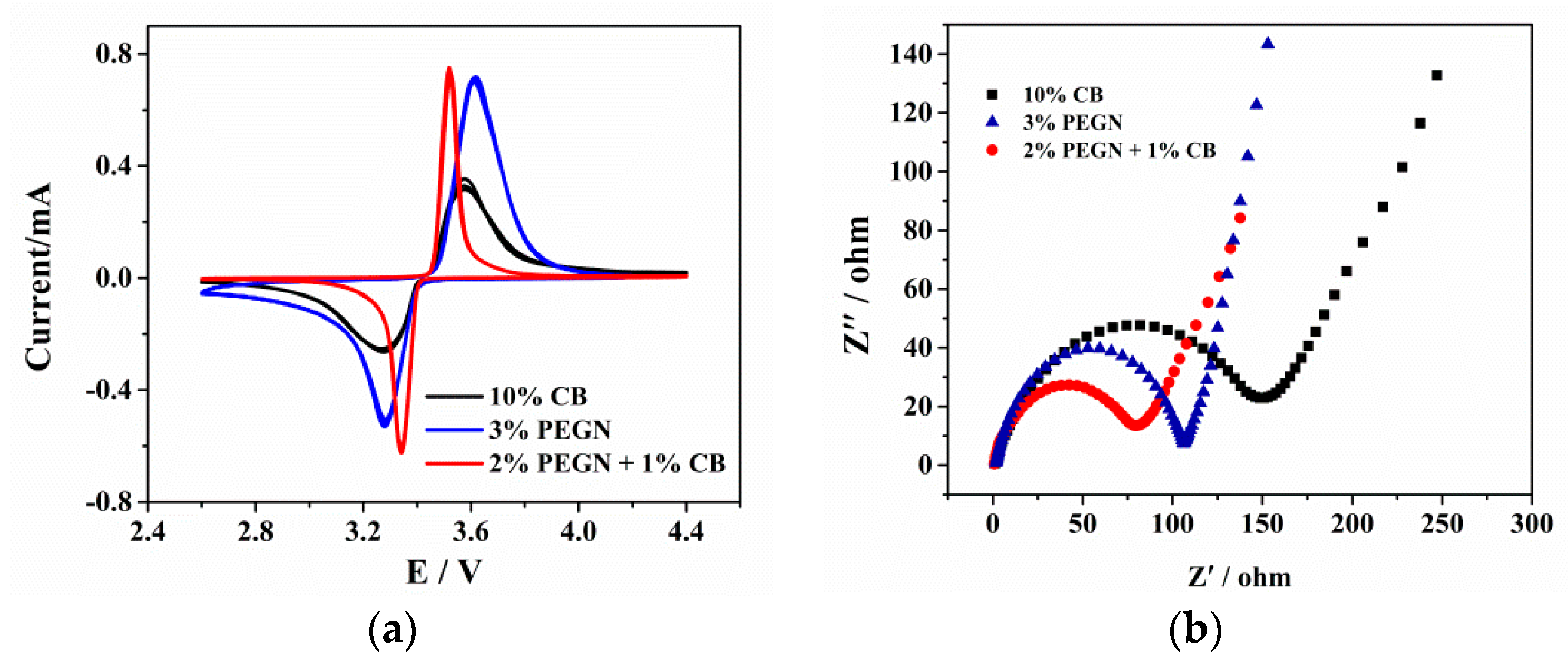
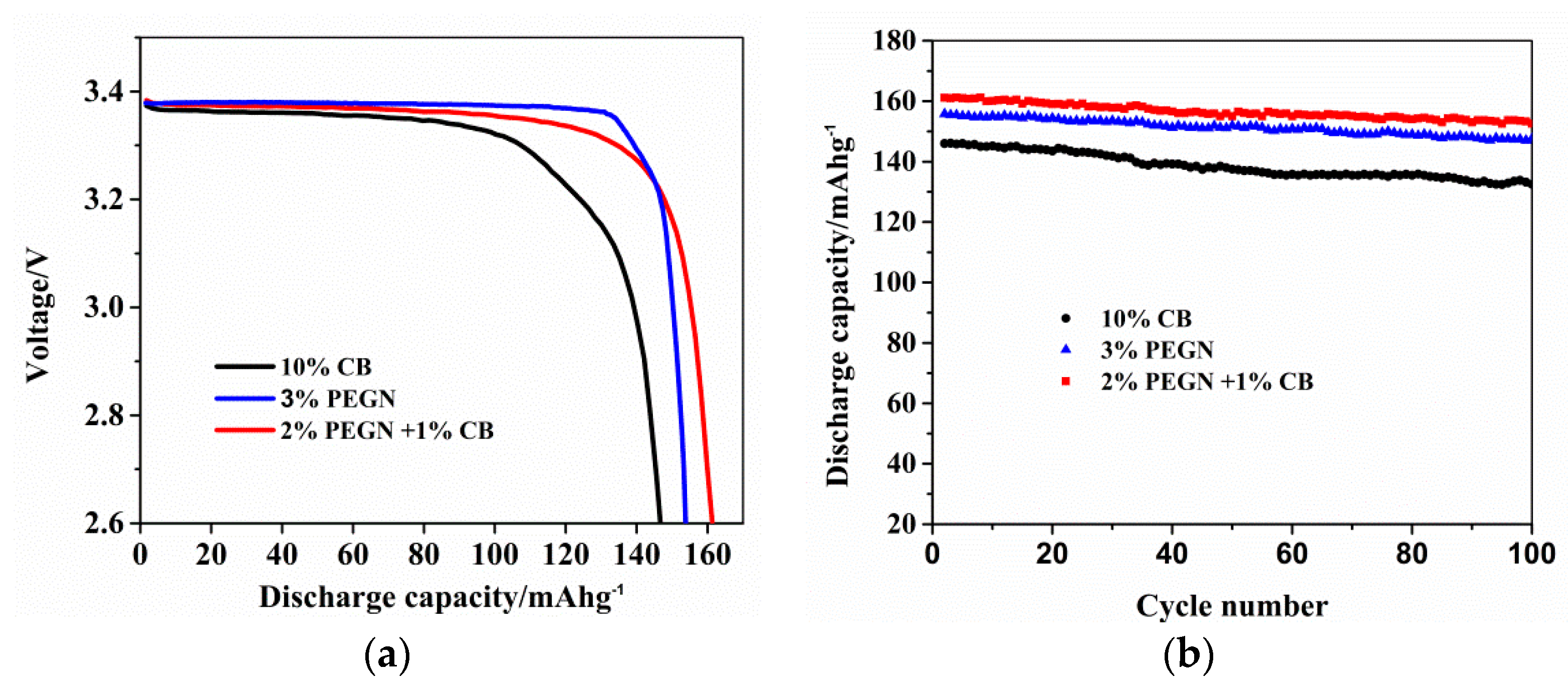
3.2. Electrochemical Properties
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, L.; Gao, P.; Gai, S.L.; He, F.; Chen, Y.J.; Zhang, M.L.; Yang, P.P. Ultra small and highly dispersed Fe3O4 nanoparticles anchored on reduced graphene for supercapacitor application. J. Electrochim. Acta 2016, 190, 566–573. [Google Scholar] [CrossRef]
- Taluja, Y.; Santhibhushan, B.; Yadav, S.; Srivastava, A. Defect and functionalized graphene for supercapacitor electrodes. Superlattices Microst. 2016, 98, 306–315. [Google Scholar] [CrossRef]
- Bae, S.; Kim, H.; Lee, Y.; Xu, X.F.; Park, J.S.; Zheng, Y.; Balakrishnan, J.; Lei, T.; Kim, H.R.; Song, Y.; et al. Roll-to-roll production of 30-inch grapheme films for transparent electrodes. Nat. Nanotechnol. 2010, 5, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tian, H.Y.; Tang, J.J.; Bai, T.; Xi, L.H.; Chen, S.M.; Zhou, X.Y. Self-assembled NiCo2O4-anchored reduced graphene oxide nanoplates as high performance anode materials for lithium ion batteries. Appl. Surf. Sci. 2017, 426, 1055–1062. [Google Scholar] [CrossRef]
- Hu, Y.; He, D.W.; Wang, Y.S.; Fu, M.; An, X.F.; Zhao, X. Defect-introduced graphene sheets with hole structure as lithium-ion battery anode. Mater. Lett. 2016, 164, 278–281. [Google Scholar] [CrossRef]
- Liu, L.J.; Huang, X.K.; Guo, X.R.; Mao, S.; Chen, J.H. Decorating in situ ultrasmall tin particles on crumpled N-doped graphene for lithium-ion batteries with a long life cycle. J. Power Sources 2016, 328, 482–491. [Google Scholar] [CrossRef]
- Jiao, J.Q.; Qiu, W.D.; Tang, J.G.; Chen, L.P.; Jing, L.Y. Synthesis of well-defined Fe3O4 nanorods/N-doped graphene for lithium-ion batteries. Nano Res. 2016, 9, 1256–1266. [Google Scholar] [CrossRef]
- Arefinia, Z.; Asgari, A. An analytical model for optimizing the performance of graphene based silicon Schottky barrier solar cells. Mater. Sci. Semicond. Proc. 2015, 35, 181–188. [Google Scholar] [CrossRef]
- Iwan, A.; Caballero-Briones, F.; Malinowski, M.; Filapek, M.; Tazbir, I.; Guerrerocontreras, J.; Kamara, S.K. Graphene oxide influence on selected properties of polymer fuel cells based on Nafion. Int. J. Hydrog. Energy 2017, 42, 15359–15369. [Google Scholar] [CrossRef]
- Jee, Y.; Karimaghaloo, A.; Macedo Andrade, A.; Moon, H.; Li, Y.; Han, J.W.; Ji, S.; Ishihara, H.; Su, P.C.; Cha, S.W.; et al. Graphene-based oxygen reduction electrodes for low temperature solid oxide fuel cells. Fuel Cells 2017, 3, 344–352. [Google Scholar] [CrossRef]
- Vallejosburgos, F.; Coudert, F.X.; Kaneko, K. Air separation with graphene mediated by nanowindow-rim concerted motion. Nat. Commun. 2018, 9, 130–135. [Google Scholar]
- Dimov, D.; Amit, I.; Gorrie, O.; Barnes, M.D.; Townsend, N.J.; Neves, I.S.; Withers, F.; Russo, S.; Craciun, M.F. Ultrahigh performance nanoengineered graphene concrete composites for multifunctional applications. Adv. Funct. Mater. 2018, 28. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Song, Z.; Li, X.; Wu, X.; Brown, N.; Naud, C.; Mayou, D.; Li, T.; Hass, J.; Marchenkov, A.N.; et al. Electronic confinement and coherence in patterned epitaxial grapheme. Science 2006, 312, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Emtsev, K.V.; Bostwick, A.; Horn, K.; Jobst, J.; Kellogg, G.L.; Ley, L.; McChesney, J.L.; Ohta, T.; Reshanov, S.A.; Röhrl, J.; et al. Towards wafer-size graphene layers by atmospheric pressure graphitization of silicon carbide. Nat. Mater. 2009, 8, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Zhao, Y.; Jang, H.; Lee, S.Y.; Kim, J.M.; Kim, K.S.; Ahn, J.H.; Kim, P.; Choi, J.Y.; Hong, B.H. Large-scale pattern growth of graphene films for stretchable transparent electrodes. Nature 2009, 457, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cai, W.; An, J.; Kim, S.; Nah, J.; Yang, D.; Piner, R.; Velamakanni, A.; Jung, I.; Tutuc, E.; et al. Large-area synthesis of high-quality and uniform graphene films on copper foils. Science 2009, 324, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Dato, A.; Radmilovic, V.; Lee, Z.; Phillips, J.; Frenklach, M. Substrate-free gas-phase synthesis of graphene sheets. Nano Lett. 2008, 8, 2012–2016. [Google Scholar] [CrossRef] [PubMed]
- Schniepp, H.C.; Li, J.L.; McAllister, M.J.; Sai, H.; Herrera-Alonso, M.; Adamson, D.H.; Prud’homme, R.K.; Car, R.; Saville, D.A.; Aksay, I.A. Functionalized single graphene sheets derived from splitting graphite oxide. J. Phys. Chem. B 2006, 110, 8535–8539. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.; Dikin, D.; Park, S.; Cai, W.W.; Mielke, L.S.; Ruoff, R.S. Characterization of thermally reduced graphene oxide by Imaging ellipsometry. J. Phys. Chem. C 2008, 112, 20264–20268. [Google Scholar] [CrossRef]
- Gilje, S.; Han, S.; Wang, M.; Wang, K.L.; Kaner, R.B. A chemical route to graphene for device applications. Nano Lett. 2007, 7, 3394–3398. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Kim, K.K.; Benayad, A.; Yoon, S.M.; Park, H.K.; Jung, I.S.; Jin, M.H.; Jeong, H.K.; Kim, J.M.; Choi, J.Y.; et al. Efficient reduction of graphite oxide by sodium borohydride and its effect on electrical conductance. Adv. Funct. Mater. 2009, 19, 1987–1992. [Google Scholar] [CrossRef]
- Yang, D.; Velamakanni, A.; Bozoklub, G.; Park, S.; Stoller, M.; Piner, R.D.; Stankovich, S.; Jung, I.; Field, D.A.; Ventrice, C.A.; et al. Chemical analysis of graphene oxide films after heat and chemical treatments by X-ray photoelectron and Micro-Raman spectroscopy. Carbon 2009, 47, 145–152. [Google Scholar] [CrossRef]
- Cote, L.J.; Cruzsilva, R.; Huang, J. Flash reduction and patterning of graphite oxide and its polymer composite. J. Am. Chem. Soc. 2009, 131, 11027–11032. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.L.; Wang, X.F.; Qian, Q.Y.; Wang, F.B.; Xia, X.H. A green approach to the synthesis of graphene nanosheets. ACS Nano 2009, 3, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- Baraket, M.; Walton, S.G.; Wei, Z.; Lock, E.H.; Robinson, J.T.; Sheehan, P. Reduction of graphene oxide by electron beam generated plasmas produced in methane/argon mixtures. Carbon 2010, 48, 3382–3390. [Google Scholar] [CrossRef]
- Teweldebrhan, D.; Balandin, A.A. Modification of graphene properties due to electron-beam irradiation. Appl. Phys. Lett. 2009, 94, 666. [Google Scholar] [CrossRef]
- Kovtyukhova, N.I.; Ollivier, P.J.; Martin, B.R.; Mallouk, T.E.; Chizhik, S.A.; Buzaneva, E.V.; Gorchinskiy, A.D. Layer-by-layer assembly of ultrathin composite films from micron-sized graphite oxide sheets and polycations. Chem. Mater. 1999, 11, 771–778. [Google Scholar] [CrossRef]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.B.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.T.; Ruoff, R.S. Graphene-based composite materials. Nature 2006, 442, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.; Diao, Y.Q.; Ding, F.; Feng, W.; Liu, X.J. Simple method for synthesizing few-layer graphene as cathodes in surface-enabled lithium ion-exchanging cells. Ionics 2016, 22, 1575–1584. [Google Scholar] [CrossRef]
- Shen, B.; Lu, D.D.; Zhai, W.T.; Zheng, W.G. Synthesis of graphene by low-temperature exfoliation and reduction of graphite oxide under ambient atmosphere. Mater. Chem. C 2013, 1, 50–53. [Google Scholar] [CrossRef]
- Meyer, J.C.; Geim, A.K.; Katsnelson, M.I.; Novoselov, K.S.; Booth, T.J.; Roth, S. The structure of suspended graphene sheets. Nature 2007, 446, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.F.; Yan, L.F.; Bangal, P.R. Preparation of graphene by the rapid and mild thermal reduction of graphene oxide induced by microwaves. Carbon 2010, 48, 1146–1152. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman spectrum of graphene and graphene layers. Phys. Rev. Lett. 2006, 97. [Google Scholar] [CrossRef] [PubMed]
- Eda, G.; Fanchini, G.; Chhowalla, M. Large-area ultrathin films of reduced graphene oxide as a transparent and flexible electronic material. Nat. Nanotechnol. 2008, 3, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; An, J.; Piner, R.D.; Jung, I.; Yang, D.X.; Velamakanni, A.; Nguyen, S.T.; Ruoff, R.S. Aqueous suspension and characterization of chemically modified graphene sheets. Chem. Mater. 2008, 20, 6592–6594. [Google Scholar] [CrossRef]
- Fan, X.B.; Peng, W.C.; Li, Y.; Li, X.Y.; Wang, S.L.; Zhang, G.L.; Zhang, F.B. Deoxygenation of exfoliated graphite oxide under alkaline conditions: A green route to graphene preparation. Adv. Mater. 2008, 20, 4490–4493. [Google Scholar] [CrossRef]
- Kim, H.J.; Jeong, H.K. Direct reform of graphite oxide electrodes by using ambient plasma for supercapacitor applications. Chem. Phys. Lett. 2017, 686, 49–54. [Google Scholar] [CrossRef]
- Yu, H.; Shang, L.; Bian, T.; Shi, R.; Waterhouse, G.I.N.; Zhao, Y.; Zhou, C.; Wu, L.Z.; Tung, C.H.; Zhang, T. Nitrogen-doped porous carbon nanosheets templated from g-C3N4 as metal-free electrocatalysts for efficient oxygen reduction reaction. Adv. Mater. 2016, 28, 5080–5086. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Bu, X.D.; Xu, L.H.; Liu, J.L.; Zhang, C. A novel LiFePO4/graphene/carbon composite as a performance-improved cathode material for lithium-ion batteries. Electrochim. Acta 2012, 64, 190–195. [Google Scholar] [CrossRef]
- Badot, J.C.; Ligneel, É.; Dubrunfaut, O.; Guyomard, D.; Lestriez, B. A multiscale description of the electronic transport within the hierarchical architecture of a composite electrode for lithium batteries. Adv. Funct. Mater. 2009, 19, 2749–2758. [Google Scholar] [CrossRef]
- Chen, Y.H.; Wang, C.W.; Liu, G.; Song, X.Y.; Battaglia, V.S.; Sastry, A.M. Selection of conductive additives in Li-ion battery cathodes-A numerical study. J. Electrochem. Soc. 2007, 154, A978–A986. [Google Scholar] [CrossRef]
- Li, Y.; Lu, X.H.; Su, F.Y. A graphene/carbon black hybrid material: A novel binary conductive additive for lithium-ion batteries. New Carbon Mater. 2015, 30, 128–132. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Z.; Li, Y.; Wang, F.; Hong, R. Plasma Exfoliated Graphene: Preparation via Rapid, Mild Thermal Reduction of Graphene Oxide and Application in Lithium Batteries. Materials 2019, 12, 707. https://doi.org/10.3390/ma12050707
Luo Z, Li Y, Wang F, Hong R. Plasma Exfoliated Graphene: Preparation via Rapid, Mild Thermal Reduction of Graphene Oxide and Application in Lithium Batteries. Materials. 2019; 12(5):707. https://doi.org/10.3390/ma12050707
Chicago/Turabian StyleLuo, Zuyun, Yuanyuan Li, Fangfang Wang, and Ruoyu Hong. 2019. "Plasma Exfoliated Graphene: Preparation via Rapid, Mild Thermal Reduction of Graphene Oxide and Application in Lithium Batteries" Materials 12, no. 5: 707. https://doi.org/10.3390/ma12050707
APA StyleLuo, Z., Li, Y., Wang, F., & Hong, R. (2019). Plasma Exfoliated Graphene: Preparation via Rapid, Mild Thermal Reduction of Graphene Oxide and Application in Lithium Batteries. Materials, 12(5), 707. https://doi.org/10.3390/ma12050707