
Plasma-Induced Catalyst Support Defects for the Photothermal Methanation of Carbon Dioxide

 , , , ,
, , , ,
Abstract
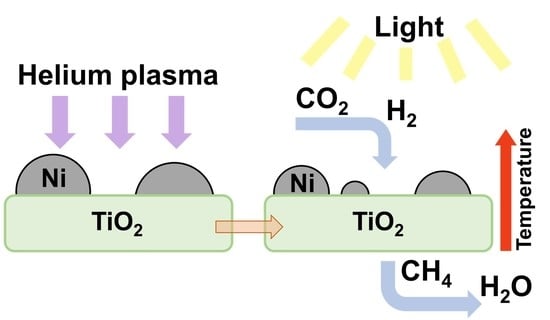
:
1. Introduction
2. Materials and Methods
2.1. Catalyst Synthesis
2.2. Catalyst Characterisation
2.3. Activity Tests
3. Results and Discussion
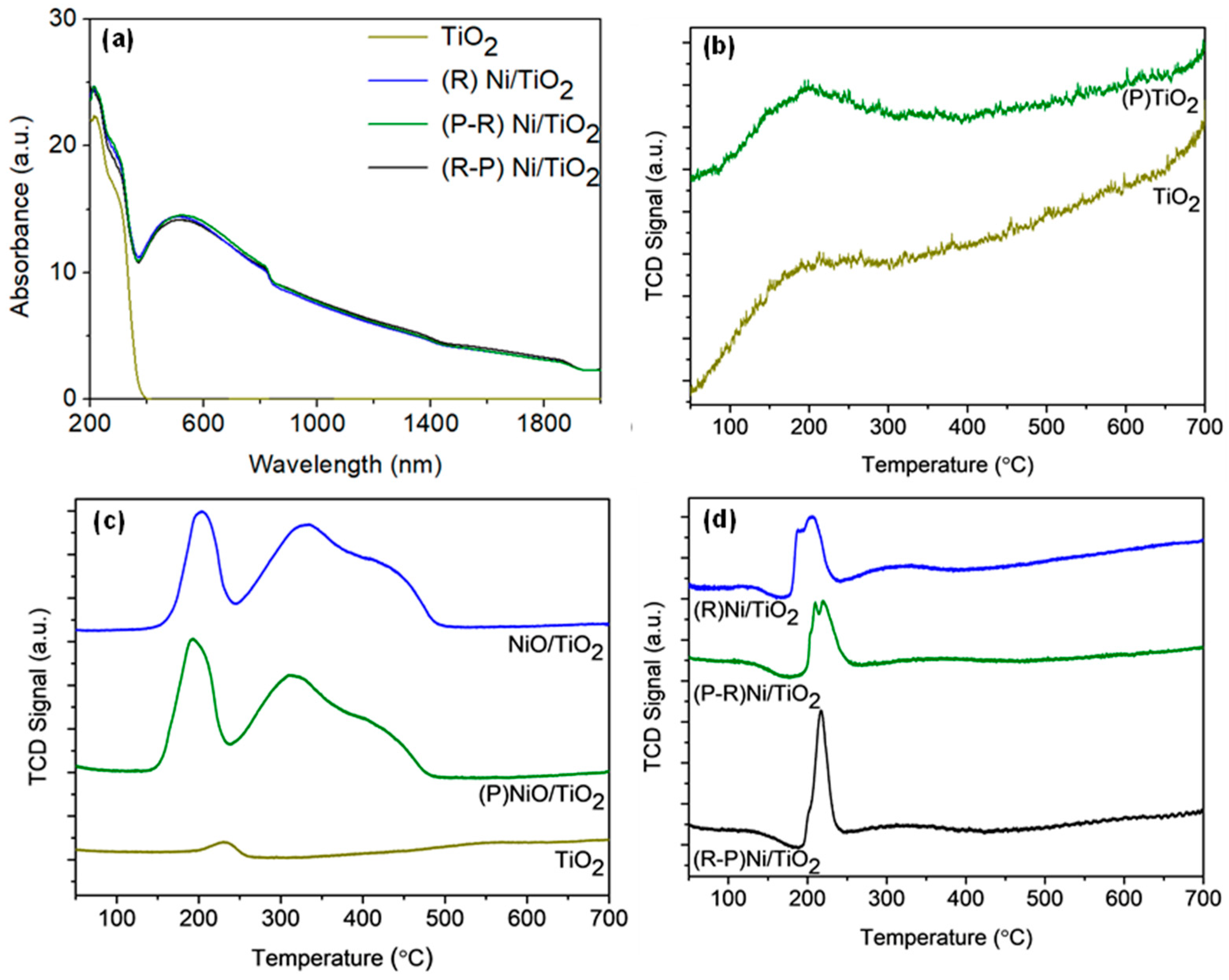
3.1. Catalyst Properties
3.2. CO2 Methanation Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Cuéllar-Franca, R.M.; Azapagic, A. Carbon capture, storage and utilisation technologies: A critical analysis and comparison of their life cycle environmental impacts. J. CO2 Util. 2015, 9, 82–102. [Google Scholar] [CrossRef]
- Ghaib, K.; Nitz, K.; Ben-Fares, F.-Z. Chemical Methanation of CO2: A Review. ChemBioEng Rev. 2016, 3, 266–275. [Google Scholar] [CrossRef]
- Mac Kinnon, M.A.; Brouwer, J.; Samuelsen, S. The role of natural gas and its infrastructure in mitigating greenhouse gas emissions, improving regional air quality, and renewable resource integration. Prog. Energy Combust. Sci. 2018, 64, 62–92. [Google Scholar] [CrossRef]
- Jia, J.; Wang, H.; Lu, Z.; O’Brien, P.G.; Ghoussoub, M.; Duchesne, P.; Zheng, Z.; Li, P.; Qiao, Q.; Wang, L.; et al. Photothermal Catalyst Engineering: Hydrogenation of Gaseous CO2 with High Activity and Tailored Selectivity. Adv. Sci. 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Wang, T.; Liu, L.; Ouyang, S.; Li, P.; Hu, H.; Kako, T.; Iwai, H.; Tanaka, A.; Ye, J. Photothermal conversion of CO2 into CH4 with H2 over Group VIII Nanocatalysts: An alternative approach for solar fuel production. Angew. Chem. Int. Ed. 2014, 4, 11662–11666. [Google Scholar] [CrossRef]
- Ren, J.; Ouyang, S.; Xu, H.; Meng, X.; Wang, T.; Wang, D.; Ye, J. Targeting Activation of CO2 and H2 over Ru-Loaded Ultrathin Layered Double Hydroxides to Achieve Efficient Photothermal CO2 Methanation in Flow-Type System. Adv. Energy Mater. 2016, 1601657. [Google Scholar] [CrossRef]
- Le, T.A.; Kang, J.K.; Park, E.D. CO and CO2 Methanation Over Ni/SiC and Ni/SiO2 Catalysts. Top. Catal. 2018, 61, 1537–1544. [Google Scholar] [CrossRef]
- Zhou, R.; Rui, N.; Fan, Z.; Liu, C. Effect of the structure of Ni/TiO2 catalyst on CO2 methanation. Int. J. Hydrog. Energy 2016, 41, 22017–22025. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Baran, R.; Dębek, R.; Motak, M.; Grzybek, T.; Gálvez, M.E.; Da Costa, P. The influence of nickel content on the performance of hydrotalcite-derived catalysts in CO2 methanation reaction. Int. J. Hydrog. Energy 2017, 42, 23548–23555. [Google Scholar] [CrossRef]
- Kesavan, J.K.; Luisetto, I.; Tuti, S.; Meneghini, C.; Iucci, G.; Battocchio, C.; Mobilio, S.; Casciardi, S.; Sisto, R. Nickel supported on YSZ: The effect of Ni particle size on the catalytic activity for CO2 methanation. J. CO2 Util. 2018, 23, 200–211. [Google Scholar] [CrossRef]
- Kim, A.; Sanchez, C.; Patriarche, G.; Ersen, O.; Moldovan, S.; Wisnet, A.; Sassoye, C.; Debecker, D.P. Selective CO2 methanation on Ru/TiO2 catalysts: Unravelling the decisive role of the TiO2 support crystal structure. Catal. Sci. Technol. 2016, 6, 8117–8128. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Wang, X.; Dong, L.; Su, T.; Li, B.; Zhou, Y.; Jiang, Y.; Luo, X.; Ji, H. CO2 methanation on Co/TiO2 catalyst: Effects of Y on the support. Chem. Eng. Sci. 2019, 210, 115245. [Google Scholar] [CrossRef]
- Low, J.; Cheng, B.; Yu, J. Surface modification and enhanced photocatalytic CO2 reduction performance of TiO2: A review. Appl. Surf. Sci. 2017, 392, 658–686. [Google Scholar] [CrossRef]
- Xu, M.; Hu, X.; Wang, S.; Yu, J.; Zhu, D.; Wang, J. Photothermal effect promoting CO2 conversion over composite photocatalyst with high graphene content. J. Catal. 2019, 377, 652–661. [Google Scholar] [CrossRef]
- Jantarang, S.; Lovell, E.C.; Tan, T.H.; Scott, J.; Amal, R. Role of support in photothermal carbon dioxide hydrogenation catalysed by Ni/CexTiyO2. Prog. Nat. Sci. Mater. Int. 2018, 28, 168–177. [Google Scholar] [CrossRef]
- Li, Z.; Liu, J.; Shi, R.; Waterhouse, G.I.N.; Wen, X.D.; Zhang, T. Fe-Based Catalysts for the Direct Photohydrogenation of CO2 to Value-Added Hydrocarbons. Adv. Energy Mater. 2021, 11, 2002783. [Google Scholar] [CrossRef]
- Batzill, M.; Morales, E.H.; Diebold, U. Influence of nitrogen doping on the defect formation and surface properties of TiO2 rutile and anatase. Phys. Rev. Lett. 2006, 96. [Google Scholar] [CrossRef]
- Pecchi, G.; Jiliberto, M.G.; Buljan, A.; Delgado, E.J. Relation between defects and catalytic activity of calcium doped LaFeO3 perovskite. Solid State Ion. 2011, 187, 27–32. [Google Scholar] [CrossRef]
- Li, Z.H.; Tian, S.X.; Wang, H.T.; Tian, H.B. Plasma treatment of Ni catalyst via a corona discharge. J. Mol. Catal. A Chem. 2004, 211, 149–153. [Google Scholar] [CrossRef]
- Liu, C.J.; Yu, K.; Zhang, Y.P.; Zhu, X.; He, F.; Eliasson, B. Characterization of plasma treated Pd/HZSM-5 catalyst for methane combustion. Appl. Catal. B Environ. 2004, 47, 95–100. [Google Scholar] [CrossRef]
- Liu, F.; Leung, Y.H.; Djurišić, A.B.; Ng, A.M.C.; Chan, W.K.; Ng, K.L.; Wong, K.S.; Liao, C.; Shih, K.; Surya, C. Effect of plasma treatment on native defects and photocatalytic activities of zinc oxide tetrapods. J. Phys. Chem. C 2014, 118, 22760–22767. [Google Scholar] [CrossRef]
- Quaas, M.; Wulff, H.; Ivanova, O.; Helm, C.A. Plasma chemical reactions of thin nickel films. Surf. Interface Anal. 2008, 40, 552–555. [Google Scholar] [CrossRef]
- Horlyck, J.; Nashira, A.; Lovell, E.; Daiyan, R.; Bedford, N.; Wei, Y.; Amal, R.; Scott, J. Plasma Treating Mixed Metal Oxides to Improve Oxidative Performance via Defect Generation. Materials 2019, 12, 2756. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Pérez, L.; Belda-Alcázar, V.; Marini, C.; Pastor-Blas, M.M.; Sepúlveda-Escribano, A.; Ramos-Fernandez, E.V. Effect of cold Ar plasma treatment on the catalytic performance of Pt/CeO2 in water-gas shift reaction (WGS). Appl. Catal. B Environ. 2018, 225, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Saputera, W.H.; Tahini, H.A.; Sabsabi, M.; Tan, T.H.; Bedford, N.M.; Lovell, E.; Cui, Y.; Hart, J.N.; Friedmann, D.; Smith, S.C.; et al. Light-Induced Synergistic Multidefect Sites on TiO2/SiO2 Composites for Catalytic Dehydrogenation. ACS Catal. 2019, 9, 2674–2684. [Google Scholar] [CrossRef]
- Tan, T.H.; Scott, J.; Ng, Y.H.; Taylor, R.A.; Aguey-Zinsou, K.F.; Amal, R. Understanding Plasmon and Band Gap Photoexcitation Effects on the Thermal-Catalytic Oxidation of Ethanol by TiO2-Supported Gold. ACS Catal. 2016, 6, 1870–1879. [Google Scholar] [CrossRef]
- Shah, M.W.; Zhu, Y.; Fan, X.; Zhao, J.; Li, Y.; Asim, S.; Wang, C. Facile Synthesis of Defective TiO2−x Nanocrystals with High Surface Area and Tailoring Bandgap for Visible-light Photocatalysis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Zhang, M.; Wang, L.; Wang, F.; Yang, L.; Li, X.; Wang, C. Plasmonic Ag-TiO2−x nanocomposites for the photocatalytic removal of NO under visible light with high selectivity: The role of oxygen vacancies. Appl. Catal. B Environ. 2017. [Google Scholar] [CrossRef]
- Kalaiarasi, S.; Sivakumar, A.; Martin Britto Dhas, S.A.; Jose, M. Shock wave induced anatase to rutile TiO2 phase transition using pressure driven shock tube. Mater. Lett. 2018, 219, 72–75. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, Y.; Zhang, J.; Pan, C. Raman spectroscopy: A new approach to measure the percentage of anatase TiO2 exposed (001) facets. J. Phys. Chem. C 2012, 116, 7515–7519. [Google Scholar] [CrossRef]
- Surmacki, J.; Wroński, P.; Szadkowska-Nicze, M.; Abramczyk, H. Raman spectroscopy of visible-light photocatalyst—Nitrogen-doped titanium dioxide generated by irradiation with electron beam. Chem. Phys. Lett. 2013, 566, 54–59. [Google Scholar] [CrossRef]
- Yu, J.; Xiang, Q.; Zhou, M. Preparation, characterization and visible-light-driven photocatalytic activity of Fe-doped titania nanorods and first-principles study for electronic structures. Appl. Catal. B Environ. 2009, 90, 595–602. [Google Scholar] [CrossRef]
- Yin, W.J.; Chen, S.; Yang, J.H.; Gong, X.G.; Yan, Y.; Wei, S.H. Effective band gap narrowing of anatase TiO2 by strain along a soft crystal direction. Appl. Phys. Lett. 2010, 96. [Google Scholar] [CrossRef] [Green Version]
- Mateo, D.; Morlanes, N.; Maity, P.; Shterk, G.; Mohammed, O.F.; Gascon, J. Efficient Visible-Light Driven Photothermal Conversion of CO2 to Methane by Nickel Nanoparticles Supported on Barium Titanate. Adv. Funct. Mater. 2021, 31. [Google Scholar] [CrossRef]
- Chen, J.; Albella, P.; Pirzadeh, Z.; Alonso-González, P.; Huth, F.; Bonetti, S.; Bonanni, V.; Åkerman, J.; Nogués, J.; Vavassori, P.; et al. Plasmonic nickel nanoantennas. Small 2011, 7, 2341–2347. [Google Scholar] [CrossRef] [Green Version]
- Nguyen Thanh, D.; Kikhtyanin, O.; Ramos, R.; Kothari, M.; Ulbrich, P.; Munshi, T.; Kubička, D. Nanosized TiO2—A promising catalyst for the aldol condensation of furfural with acetone in biomass upgrading. Catal. Today 2016, 277, 97–107. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, H.; Andino, J.M.; Li, Y. Photocatalytic CO2 reduction with H2O on TiO2 nanocrystals: Comparison of anatase, rutile, and brookite polymorphs and exploration of surface chemistry. ACS Catal. 2012, 2, 1817–1828. [Google Scholar] [CrossRef]
- Inturi, S.N.R.; Suidan, M.; Smirniotis, P.G. Influence of synthesis method on leaching of the Cr-TiO2 catalyst for visible light liquid phase photocatalysis and their stability. Appl. Catal. B Environ. 2016, 180, 351–361. [Google Scholar] [CrossRef]
- Meeyoo, V.; Panchan, N.; Phongprueksathat, N.; Traitangwong, A.; Guo, X.; Li, C.; Rirksomboon, T. Low Temperature Methanation of CO2 on High Ni Content Ni-Ce-ZrOδ Catalysts Prepared via One-Pot Hydrothermal Synthesis. Catalysts 2019, 10, 32. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Lua, A.C. Sol-gel synthesis of Ni and Ni supported catalysts for hydrogen production by methane decomposition. RSC Adv. 2014, 4, 42159–42167. [Google Scholar] [CrossRef]
- Grosvenor, A.P.; Biesinger, M.C.; Smart, R.C., St.; McIntyre, N.S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 2006, 600, 1771–1779. [Google Scholar] [CrossRef]
- An, H.R.; Park, S.Y.; Kim, H.; Lee, C.Y.; Choi, S.; Lee, S.C.; Seo, S.; Park, E.C.; Oh, Y.K.; Song, C.G.; et al. Advanced nanoporous TiO2 photocatalysts by hydrogen plasma for efficient solar-light photocatalytic application. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.L.; Poznyak, T.; Valenzuela, M.A.; Tiznado, H.; Chairez, I. Surface interactions and mechanistic studies of 2,4-dichlorophenoxyacetic acid degradation by catalytic ozonation in presence of Ni/TiO2. Chem. Eng. J. 2013, 222, 426–434. [Google Scholar] [CrossRef]
- Tahir, M.; Tahir, B.; Amin, N.A.S.; Muhammad, A. Photocatalytic CO2 methanation over NiO/In2O3 promoted TiO2 nanocatalysts using H2O and/or H2 reductants. Energy Convers. Manag. 2016, 119, 368–378. [Google Scholar] [CrossRef]
- Bharti, B.; Kumar, S.; Lee, H.N.; Kumar, R. Formation of oxygen vacancies and Ti3+ state in TiO2 thin film and enhanced optical properties by air plasma treatment. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Li, J.; Ye, Y.; Ye, L.; Su, F.; Ma, Z.; Huang, J.; Xie, H.; Dorokin, D.E.; Zimina, A.; Grunwaldt, J.-D.; et al. Sunlight induced photo-thermal synergistic catalytic CO2 conversion via localized surface plasmon resonance of MoO3−x. J. Mater. Chem. A 2019, 7, 2821–2830. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Wang, B.; Sun, Y.; Wang, X.; Fu, M.; Wu, J.; Chen, L.; Tan, Y.; Ye, D. Plasma-Assisted Surface Interactions of Pt/CeO2 Catalyst for Enhanced Toluene Catalytic Oxidation Bingxu. Catalysts 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Pretreatment a | SBET (m2/g) b | TiO2 Crystal Size (nm) c | NiO Crystal Size (nm) c | Ni Crystal Size (nm) c |
|---|---|---|---|---|---|
| TiO2 | N/A | 104 | 22.0 | N/A | N/A |
| NiO/TiO2 | N/A | 66 | 21.0 | 7.7 | N/A |
| (R)Ni/TiO2 | Reduced | N.D. | 23.6 | N/A | 13.1 |
| (P)NiO/TiO2 | Plasma treated | 75 | 21.0 | 9.3 | N/A |
| (P-R)Ni/TiO2 | Plasma treated then reduced | N.D. | 22.6 | N/A | 13.7 |
| (R-P)Ni/TiO2 | Reduced then plasma treated | N.D. | 23.0 | N/A | 12.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jantarang, S.; Ligori, S.; Horlyck, J.; Lovell, E.C.; Tan, T.H.; Xie, B.; Amal, R.; Scott, J. Plasma-Induced Catalyst Support Defects for the Photothermal Methanation of Carbon Dioxide. Materials 2021, 14, 4195. https://doi.org/10.3390/ma14154195
Jantarang S, Ligori S, Horlyck J, Lovell EC, Tan TH, Xie B, Amal R, Scott J. Plasma-Induced Catalyst Support Defects for the Photothermal Methanation of Carbon Dioxide. Materials. 2021; 14(15):4195. https://doi.org/10.3390/ma14154195
Chicago/Turabian StyleJantarang, Salina, Simone Ligori, Jonathan Horlyck, Emma C. Lovell, Tze Hao Tan, Bingqiao Xie, Rose Amal, and Jason Scott. 2021. "Plasma-Induced Catalyst Support Defects for the Photothermal Methanation of Carbon Dioxide" Materials 14, no. 15: 4195. https://doi.org/10.3390/ma14154195