
Electropolymerization of Metallo-Octaethylporphyrins: A Study to Explore Their Sensing Capabilities



Abstract
:1. Introduction
2. Materials and Methods
Analytical Performance
3. Results and Discussion
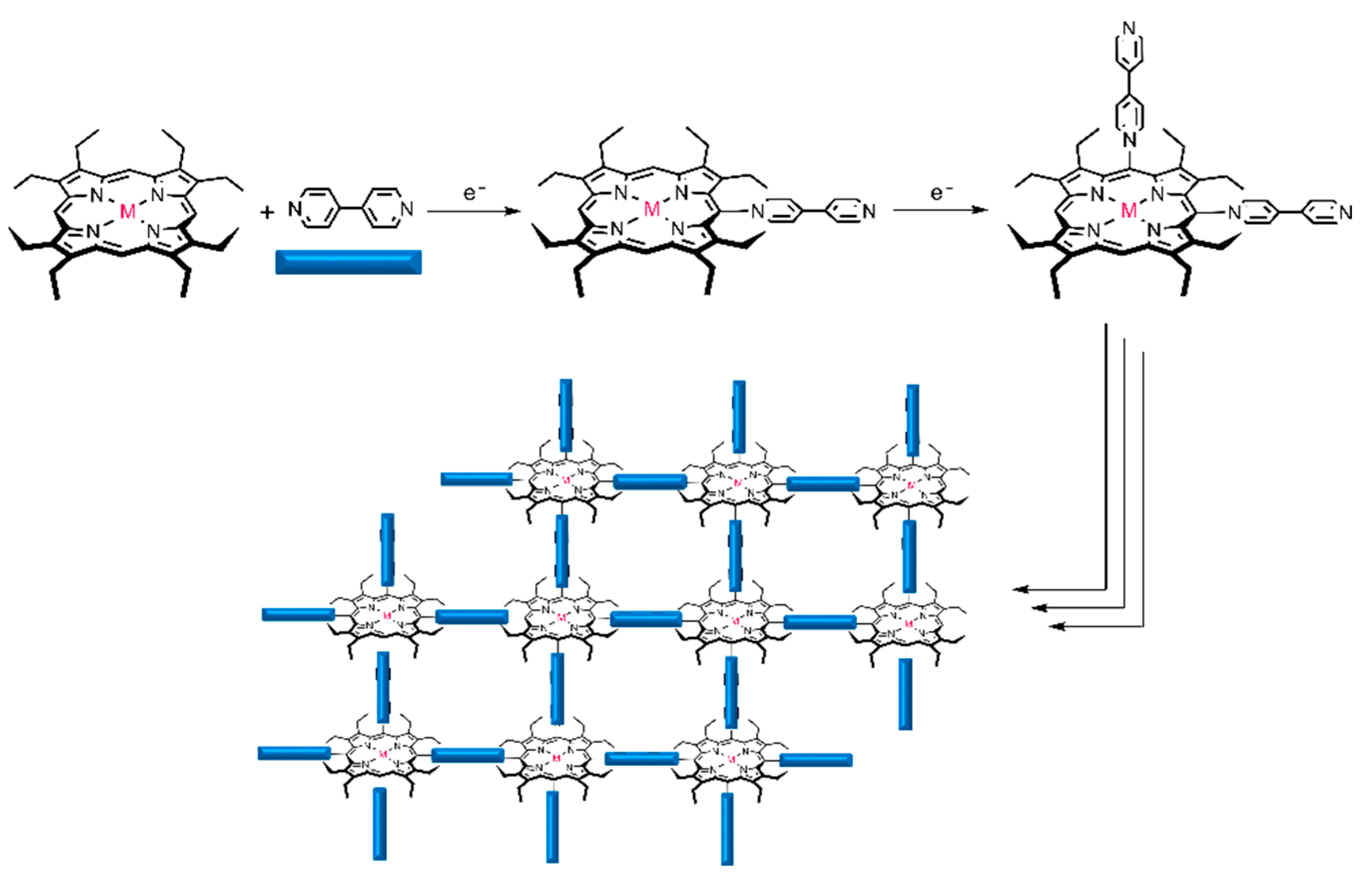
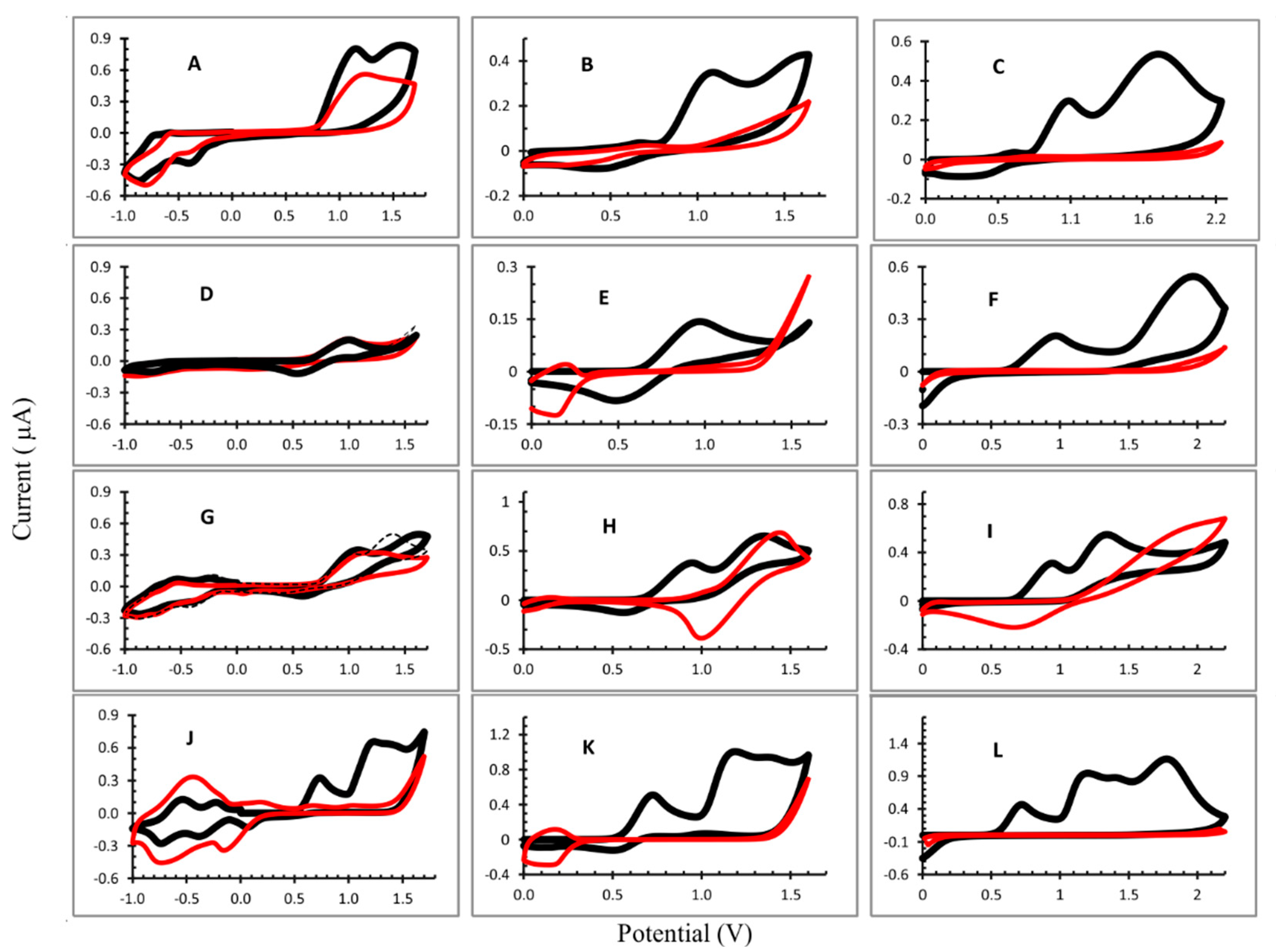
3.1. Electropolymerization
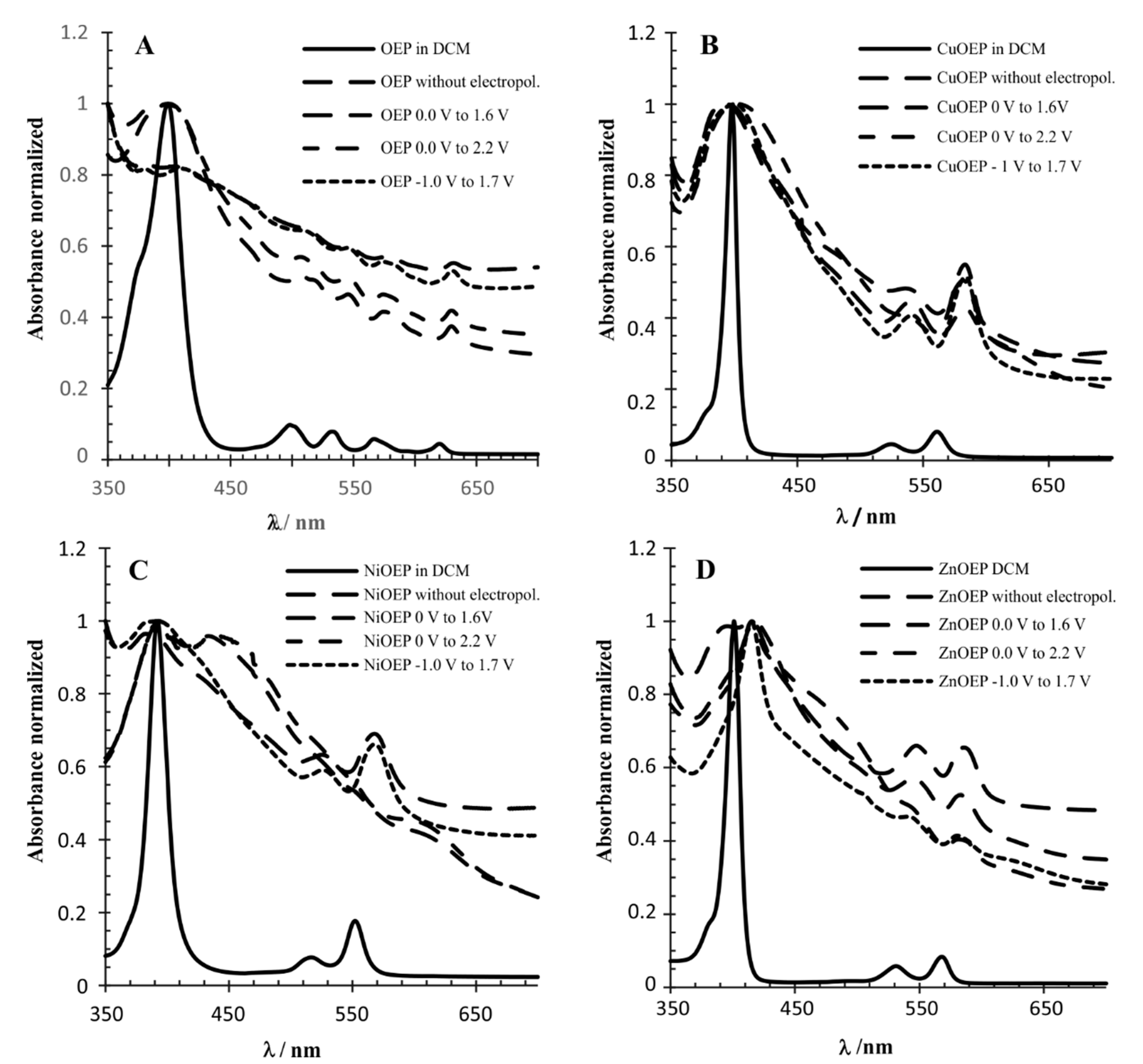
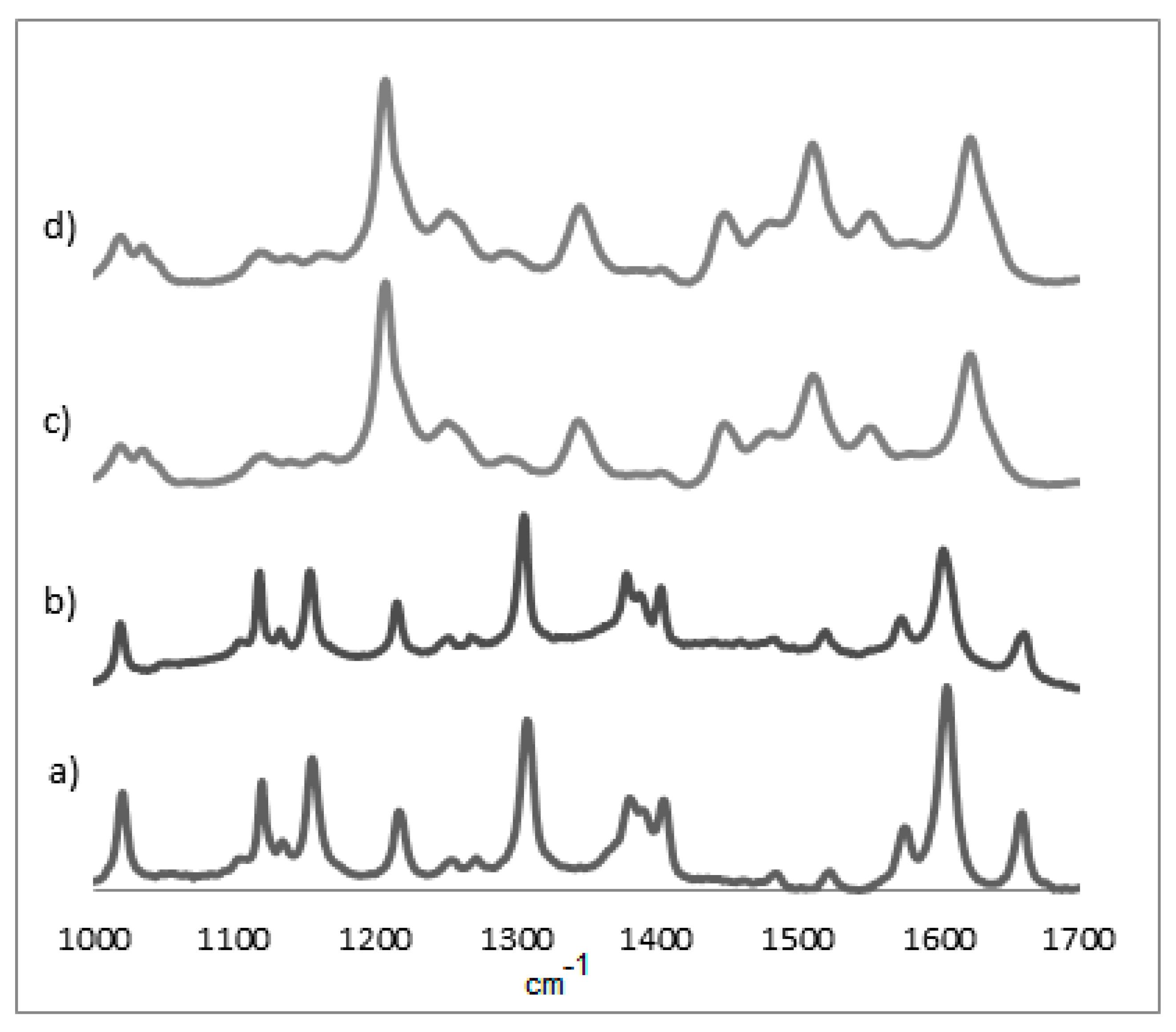
3.2. UV-Vis and Raman Characterization
3.3. Analytical Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) The Porphyrin Handbook; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Holten, D.; Bocian, D.F.; Lindsey, J.S. Probing Electronic Communication in Covalently Linked Multiporphyrin Arrays. A Guide to the Rational Design of Molecular Photonic Devices. Acc. Chem. Res. 2002, 35, 57–69. [Google Scholar]
- Imahori, H.; Mitamura, K.; Shibano, Y.; Umeyama, T.; Matano, Y.; Yoshida, K.; Isoda, S.; Araki, Y.; Ito, O. A Photoelectrochemical Device with a Nanostructured SnO2 Electrode Modified with Composite Clusters of Porphyrin-Modified Silica Nanoparticle and Fullerene. J. Phys. Chem. B 2006, 110, 11399–11405. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.J.; Cho, Y.; Min, S.K.; Kim, W.Y.; Kim, K.S. Chromium Porphyrin Arrays As Spintronic Devices. J. Am. Chem. Soc. 2011, 133, 9364–9369. [Google Scholar] [CrossRef] [PubMed]
- Picot, M.; Nicolas, I.; Poriel, C.; Rault-Berthelot, J.; Barrière, F. On the nature of the electrode surface modification by cathodic reduction of tetraarylporphyrin diazonium salts in aqueous media. Electrochem. Commun. 2012, 20, 167–170. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Brückner, C.; Li, C.M.; Lei, Y. Poly[meso-tetrakis(2-thienyl)porphyrin] for the sensitive electrochemical detection of explosives. Sens. Actuators B Chem. 2010, 147, 191–197. [Google Scholar] [CrossRef]
- Carballo, R.; Rinaldi, A.L.; Rezzano, I.N. Electrochemical study of azide bridged heterobimetallic films of porphyrins: Application as an impedimetric sensor. Electrochim. Acta 2016, 222, 1700–1708. [Google Scholar] [CrossRef]
- Macor, K.A.; Spiro, T.G. Porphyrin electrode films prepared by electrooxidation of metalloprotoporphyrins. J. Am. Chem. Soc. 1983, 105, 5601–5607. [Google Scholar] [CrossRef]
- Bedioui, F.; Devynck, J.; Bied-Charreton, C. Immobilization of metalloporphyrins in electropolymerized films: Design and applications. Acc. Chem. Res. 1995, 28, 30–36. [Google Scholar] [CrossRef]
- Carvalho de Medeiros, M.A.; Gorgy, K.; Deronzier, A.; Cosnier, S. Design of new electropolymerized polypyrrole films of polyfluorinated Zn(II) and Mn(III) porphyrins: Towards electrochemical sensors. Mater. Sci. Eng. C 2008, 28, 731–738. [Google Scholar] [CrossRef]
- Schaming, D.; Ahmed, I.; Hao, J.; Alain-Rizzo, V.; Farha, R.; Goldmann, M.; Xu, H.; Giraudeau, A.; Audebert, P.; Ruhlmann, L. Easy methods for the electropolymerization of porphyrins based on the oxidation of the macrocycles. Electrochim. Acta 2011, 56, 10454–10463. [Google Scholar] [CrossRef]
- Staden, R.-I.S.-v.; van Staden, J.F.; Balasoiu, S.C. Disposable Stochastic Dot Sensors for the Assay of Ascorbic Acid in Pharmaceutical Samples, Beverages, and Biological Fluids. Anal. Lett. 2011, 44, 2280–2286. [Google Scholar] [CrossRef]
- Hao, J.; Giraudeau, A.; Ping, Z.; Ruhlmann, L. Supramolecular Assemblies Obtained by Large Counteranion Incorporation in a Well-Oriented Polycationic Copolymer. Langmuir 2008, 24, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Leroux, Y.; Schaming, D.; Ruhlmann, L.; Hapiot, P. SECM Investigations of Immobilized Porphyrins Films. Langmuir 2010, 26, 14983–14989. [Google Scholar] [CrossRef] [PubMed]
- Gil-Ramírez, G.; Karlen, S.D.; Shundo, A.; Porfyrakis, K.; Ito, Y.; Briggs, G.A.D.; Morton, J.J.L.; Anderson, H.L. A Cyclic Porphyrin Trimer as a Receptor for Fullerenes. Org. Lett. 2010, 12, 3544–3547. [Google Scholar] [CrossRef]
- Davis, N.K.S.; Thompson, A.L.; Anderson, H.L. A Porphyrin Fused to Four Anthracenes. J. Am. Chem. Soc. 2011, 133, 30–31. [Google Scholar] [CrossRef]
- Giraudeau, A.; Schaming, D.; Hao, J.; Farha, R.; Goldmann, M.; Ruhlmann, L. A simple way for the electropolymerization of porphyrins. J. Electroanal. Chem. 2010, 638, 70–75. [Google Scholar] [CrossRef]
- Kadish, K.M.; Van Caemelbecke, E.; Boulas, P.; D’Souza, F.; Vogel, E.; Kisters, M.; Medforth, C.J.; Smith, K.M. First reversible electrogeneration of triply oxidized nickel porphyrins and porphycenes. Formation of nickel(III) .pi. dications. Inorg. Chem. 1993, 32, 4177–4178. [Google Scholar] [CrossRef]
- Giraudeau, A.; Ruhlmann, L.; El Kahef, L.; Gross, M. Electrosynthesis and Characterization of Symmetrical and Unsymmetrical Linear Porphyrin Dimers and Their Precursor Monomers. J. Am. Chem. Soc. 1996, 118, 2969–2979. [Google Scholar] [CrossRef]
- Huo, Z.; Azcarate, I.; Farha, R.; Goldmann, M.; Xu, H.; Hasenknopf, B.; Lacôte, E.; Ruhlmann, L. Copolymeric films obtained by electropolymerization of porphyrins and dipyridyl-spacers including Dawson-type polyoxometalates. J. Solid State Electrochem. 2015, 19, 2611–2621. [Google Scholar] [CrossRef]
- Nahor, G.S.; Neta, P.; Hambright, P.; Robinson, L.R. One-electron oxidation of nickel porphyrins: Effect of structure and medium on formation of nickel(III) porphyrin or nickel(II) porphyrin.pi.-radical cation. J. Phys. Chem. 1991, 95, 4415–4418. [Google Scholar] [CrossRef]
- Seth, J.; Palaniappan, V.; Bocian, D.F. Oxidation of Nickel(II) Tetraphenylporphyrin Revisited. Characterization of Stable Nickel(III) Complexes at Room Temperature. Inorg. Chem. 1995, 34, 2201–2206. [Google Scholar] [CrossRef]
- Terekhov, S.N.; Shulga, A.M. Resonance Raman spectra and structure of meso-monosubstituted metalloporphyrins. J. Mol. Struct. 1999, 484, 63–73. [Google Scholar] [CrossRef]
- Ruhlmann, L.; Schulz, A.; Giraudeau, A.; Messerschmidt, C.; Fuhrhop, J.-H. A Polycationic Zinc-5,15-dichlorooctaethylporphyrinate-viologen Wire. J. Am. Chem. Soc. 1999, 121, 6664–6667. [Google Scholar] [CrossRef]
- Long, G.L.; Winefordner, J.D. Limit of Detection A Closer Look at the IUPAC Definition. Anal. Chem. 1983, 55, 712A–724A. [Google Scholar]
- Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation; US Department of Health and Human Services, FDA, Center for Drug Evaluation and Research: Rockville, MD, USA, 2018. [Google Scholar]
- University of Iowa Hospitals and Clinics. Phenobarbital. Available online: https://uihc.org/health-topics/phenobarbital#:~:text=The%20blood%20level%20of%20phenobarbital,between%2010%2D40%C2%B5g%2Fml (accessed on 16 September 2022).
- The Royal Children’s Hospital Melbourne. Phenobarbitone Poisoning. Available online: https://www.rch.org.au/clinicalguide/guideline_index/Phenobarbitone_poisoning/ (accessed on 16 September 2022).
- Samadi, A.; Khoubnasabjafari, M.; Barzegar, M.; Sadeghvand, S.; Shiva, S.; Jouyban, A. Simultaneous Determination of Phenobarbital, Phenytoin, Carbamazepine and Carbamazepine-10,11-epoxide in Plasma of Epileptic Patients. Pharm. Sci. 2019, 25, 345–351. [Google Scholar] [CrossRef]
- Peres, M.D.; Nascimento, S.; Pelição, F.S. A new clean-up approach for stomach content toxicological analysis. Forensic Sci. Int. 2019, 302, 109936. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxidation (V) | Reduction (V) | |||
|---|---|---|---|---|
| Polymer | Dication | Radical Cation | Radical Anion | |
| OEP 1 | 1.56 | 1.15 | −0.397; −0.813 | |
| OEP 2 | 1.59 | 1.05 | ||
| OEP 3 | 1.70 | 1.05 | ||
| CuOEP 1 | 1.00 | 0.535 | −0.349; −0.781 | |
| CuOEP 2 | 0.972 | 0.486 | 0.137 | |
| CuOEP 3 | 1.968 | 0.972 | ||
| NiOEP 1 | 1.367 | 1.096 | 0.586 | −0.427; −0.872 |
| NiOEP 2 | 1.353 | 0.947 | 0.559 | |
| NiOEP 3 | 1.343 | 0.945 | ||
| ZnOEP 1 | 1.235 | 0.732 | 0.061; −0.388; −0.737 | |
| ZnOEP 2 | 1.189 | 0.725 | ||
| ZnOEP 3 | 1.198 * 1.775 | 0.720 | ||
| λmax (nm) | ||
|---|---|---|
| Modified Electrode | Soret Band | Q Bands |
| OEP (DCM) | 399 | 498; 533; 567; 620 |
| OEP * | 381; 404 | 512.5 545; 574; 632 |
| OEP 1 | 380; 410 | 513; 545; 574; 631 |
| OEP 2 | 403 | 509; 546; 575; 630 |
| OEP 3 | 399 | 507; 545; 574; 630 |
| CuOEP (DCM) | 398 | 525; 561 |
| CuOEP * | 393 | 541; 584 |
| CuOEP 1 | 400 | 541, 584 |
| CuOEP 2 | 394 | 536; 582 |
| CuOEP 3 | 403 | 584 |
| NiOEP (DCM) | 392 | 516; 552 |
| NiOEP * | 382.5 | 525; 568 |
| NiOEP 1 | 391 | 526; 568 |
| NiOEP 2 | 393; 434 | 557; 616 |
| NiOEP 3 | 393; 445 | 554, 614 |
| ZnOEP (DCM) | 401 | 531; 568 |
| ZnOEP * | 394; 415 | 548; 586 |
| ZnOEP 1 | 416 | 543; 579 |
| ZnOEP 2 | 418 | 546; 583 |
| ZnOEP 3 | 416 | 582 |
| Film | ν21 | ν4 | ν3 | ν2 | ν10 |
|---|---|---|---|---|---|
| OEP * | 1354 | - | - | ||
| OEP 1 | 1361 | 1541 | 1582 | ||
| OEP 2 | 1362 | 1539 | 1581 | ||
| OEP 3 | 1360 | 1540 | 1579 | ||
| CuOEP * | 1310 | 1373 | 1499 | 1568 1580 | 1637 |
| CuOEP 1 | 1312 | 1375 | 1502 | 1569 1584 | 1640 |
| CuOEP 2 | 1313 | 1376 | 1514 | 1569 1585 | 1640 |
| CuOEP 3 | 1311 | 1376 | 1504 | 1565 1581 | 1634 |
| NiOEP * | 1380 | 1522 | 1575 | ||
| NiOEP 1 | 1378 | 1519 | 1573 | ||
| NiOEP 2 | 1344 | 1511 | 1548 | ||
| NiOEP 3 | 1345 | 1510 | 1551 | ||
| ZnOEP * | 1353 | - | - | ||
| ZnOEP 1 | 1347 | 1513 | 1551 | ||
| ZnOEP 2 | 1346 | 1513 | 1551 | ||
| ZnOEP 3 | 1347 | 1512 | 1551 |
| Parameter | Value | |
|---|---|---|
| Linear range | 10–150 mg L−1 (43.07–646 µM) | |
| R2 | 0.9957 | |
| LOD | 10 mg L−1 (43.07 µM) | |
| LOQ | 35 mg L−1 (150.73 µM) | |
| PB conc. (mg/L) | Accuracy (% Recovery) | Precision (% RSD) |
| 30 (129.2 µM) | 118.10 | 3.48 |
| 60 (258.4 µM) | 93.81 | 4.41 |
| 118 (508 µM) | 91.93 | 5.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nascentes, C.C.; Aguilar, I.; Gil-Ramírez, G.; Gonzalez-Rodriguez, J. Electropolymerization of Metallo-Octaethylporphyrins: A Study to Explore Their Sensing Capabilities. Materials 2022, 15, 6598. https://doi.org/10.3390/ma15196598
Nascentes CC, Aguilar I, Gil-Ramírez G, Gonzalez-Rodriguez J. Electropolymerization of Metallo-Octaethylporphyrins: A Study to Explore Their Sensing Capabilities. Materials. 2022; 15(19):6598. https://doi.org/10.3390/ma15196598
Chicago/Turabian StyleNascentes, Clésia C., Ivette Aguilar, Guzmán Gil-Ramírez, and Jose Gonzalez-Rodriguez. 2022. "Electropolymerization of Metallo-Octaethylporphyrins: A Study to Explore Their Sensing Capabilities" Materials 15, no. 19: 6598. https://doi.org/10.3390/ma15196598