1. Introduction
Open-cell semi-rigid polyurethane (PUR) foams are used as thermal insulation materials. Their features are low thermal conductivity coefficient (36–42 mW/m∙K), low apparent density (8–15 kg/m
3), high water vapour permeability and resistance to mould and mildew. Open-cell PUR foams are sprayed directly onto an uninsulated surface using a high-pressure application device [
1,
2,
3]. The main application of open-cell spray PUR foams is the thermal insulation of attics, ceilings and interior walls [
4].
PUR materials, including PUR foams, are mainly synthesized using raw materials of petrochemical origin. The global market for polyols was approximately USD 26 billion in 2019 and is expected to have grown to USD 34 billion by 2024 [
5]. Increasing the sustainability of PUR through the use of renewable or waste-based raw materials has been a topic of research for years. Currently, a significant shift toward bio-based polyurethanes is also being observed across the PUR industry [
5].
Renewable raw materials such as vegetable oils are mainly used to synthesize bio-based PUR [
6,
7], although other raw materials such as microalgae oils, lignin and polysaccharides are also being studied [
5,
8]. Waste raw materials such as tall oil (a waste from the cellulose industry) [
9], waste poly(ethylene terephthalate) and used cooking oils are also being intensively studied [
10,
11].
The most common vegetable oils used to synthesize bio-polyols are rapeseed oil, sunflower oil, palm oil and soybean oil [
5,
6]. However, edible oils are important components of the human diet and their use in the chemical industry is controversial. Waste oils and non-edible oils can be alternatives to edible oils in chemical syntheses [
12,
13].
A non-edible oil used in the chemical industry to synthesize bio-polyols is castor oil. Unmodified castor oil contains hydroxyl groups in its structure and so can react with isocyanate groups to form a urethane bond. Disadvantages of castor oil are its low functionality and the presence of secondary hydroxyl groups that have lower reactivity compared to primary groups. Therefore, in order to improve its properties and broaden the applicability, it is necessary to subject the oil to chemical modifications, as in the case of other vegetable oils [
6].
Used cooking oils (UCOs) also find application in the synthesis of bio-polyols. Studies have shown that changes in the chemical structure of UCOs have a negligible impact on the synthesis process and properties of bio-polyols [
14]. A disadvantage of UCO in general can be its heterogeneous composition, which can be a mixture of many sorts of oils, as well as animal fats and impurities [
15].
Another raw material with a great potential for use in chemical syntheses is non-edible oil from oilseed radish (OR) (
Raphanus sativus (L.) var.
Oleiferus) [
16,
17,
18,
19,
20]. OR is characterized by a high content of erucic acid containing one double bond located in the 13th position along the fatty acid chain. The iodine value of crude OR is approximately 100–110 g I
2/100 g, which is similar to that of rapeseed oil. Kim et al. [
16] studied the epoxidation of OR with the use of peracetic acid. The conversion of OR to epoxidized oil was 69% and the selectivity of the process was 85%. It was found that epoxidation of oils with higher unsaturated bond contents (linseed oil, soybean oil and cottonseed oil) was characterized by higher conversion and selectivity. The use of OR as a raw material for the synthesis of bio-based PUR has not been described in the literature yet.
Another oilseed crop with high potential in the chemical industry is industrial hemp (Cannabis
sativa L.). Nearly all parts of the hemp plant, including the fibres, seeds and inflorescence, find an industrial use [
21]. Hemp seed oil (HSO) belongs to edible oils, but because of its low smoke point its main application is not in the food but in the cosmetics industry (shower gels, shampoos, hair and body balms, day and night creams and others) [
22].
New varieties of industrial oil-type hemp are characterized by high seed yields with low growth of vegetative parts and a well-developed inflorescence, as well as a shorter growing period than in typical fibre hemp. The first registered variety of oil-type hemp is ″Henola″ grown at the Institute of Natural Fibres and Medicinal Plants (Poznań, Poland) with an average oil content of approximately 33 wt. % in dry seed weight [
23]. HSO is characterized by a very high content of linoleic acid with two unsaturated bonds (C18:2, 55–60%), and linolenic acid with three unsaturated bonds (C18:3, 17–35%). The iodine value of HSO ranges from 140 to 175 g I
2/100 g [
24] and is higher than that of soybean oil (128–143 g I
2/100 g), sunflower oil (110–143 g I
2/100 g), rapeseed oil (110–126 g I
2/100 g) and palm oil (44–58 g I
2/100 g) [
25]. Epoxidized HSOs are being investigated as raw materials for the synthesis of acrylated bio-polymers used mainly as a coating material for plastic, paper and wood [
26], as well as for the synthesis of bio-based epoxy resins [
24,
27]. Surender et al. [
28] synthesized hydroxyl derivatives of HSO by subjecting the oil to epoxidation and ring-opening reactions with various agents: water, ethanol and butanol. The products had hydroxyl values of 131, 120, 129 mgKOH/g, respectively, and were used to synthesize thermoplastic PUR elastomers.
Here, we detail the development of open-cell PUR foams in which the polyol component was entirely bio-polyol synthesized from various oils: used cooking oil, rapeseed oil, oilseed radish oil and hemp seed oil. The selected raw materials are representatives of four groups of oils: edible oils, non-edible oils, waste oils and oils that are not a primary source of lipids. In the literature, there is no report on the synthesis of PUR foams containing bio-polyols from oil radish oil and hemp seed oil.
Table 1 shows the fatty acid compositions of the oils used in the study based on the literature data.
Figure 1 shows the chemical structure of fatty acids.
2. Materials and Methods
2.1. Plant Materials
Seeds of oilseed radish (
Raphanus sativus L. var.
oleiformis) and rapeseed (
Brassica napus L. var.
napus) were purchased from FN Granum (Wodzierady, Poland); industrial hemp (
Cannabis sativa L. var.
sativa) was purchased from Nutrilla (Węgrzce, Poland). Used cooking oil (UCO) was supplied by local restaurants. Vegetable oils were extruded using a Wartmann (Tilburg, The Netherland) WM-2002OP miniature extruder. The oils were not subjected to any chemical refining process, only decanted from the sediment.
Figure 2 shows the oilseeds used.
Figure 3 shows the crude oils and UCO.
2.2. Reagents
Glacial acetic acid (GAA) (99.5 wt %), hydrogen peroxide (HP) (30 wt %) and diethylene glycol (DEG) were supplied by Avantor Performance Materials Poland (Gliwice, Poland). Ion exchange resin Amberlite® IR 120 H and tetrafluoroboric acid (48% wt. % in water) were provided by Sigma-Aldrich (Darmstadt, Germany). Polymeric methylene-4,4′-diphenyl diisocyanate (pMDI) Ongronat® 2100 was supplied by BorsodChem (Kazincbarcika, Hungary). Polycat® 15 (non-emissive balanced amine catalyst), Polycat® 140 (reactive catalyst with high selectivity towards the water-isocyanate blowing reaction), KOSMOS® 19 (gelling catalyst), TEGOSTAB® B 8870 (polysiloxane polyether surfactant used in high-water formulations), ORTEGOL® 500 (cell-opener) and Dabco® EM400 (emulsifier used in water-blown, low-density spray polyurethane foam) were supplied by Evonik Industries AG (Essen, Germany). TCPP (tris (1-chloro-2-propyl) phosphate) used as a flame retardant was purchased from Lanxess AG (Cologne, Germany).
2.3. Characterization of Oils and Their Derivatives
Vegetable oils, epoxidized oils and bio-polyols were subjected to analytical, physicochemical and spectroscopic analyses.
Iodine value (Ival) determining the content of unsaturated bonds was investigated in accordance with the PN-87/C-04281 standard and calculated by using Equation (1).
where
Vb and
Vs are volumes in mL of sodium thiosulfate solution used for the blank and sample, respectively;
Ct is the concentration of sodium thiosulfate solution in mol/dm
3;
ms is the mass of the sample in g.
Hydroxyl value (Hval) was determined in accordance with the PN-C-89052-03:1993 standard. In this method, the hydroxyl groups undergo acetylation using acetic anhydride. The excess of the acetic anhydride is decomposed by a water addition and followed by titration using a solution of potassium hydroxide in the presence of thymolphthalein as an indicator. Hval was calculated by using Equation (2)
where
Vb and
Vs are volumes in ml of potassium hydroxide used for the blank and sample, respectively;
CKOH is the concentration of potassium hydroxide solution in mol/dm
3;
ms is the mass of the sample in g.
Acid value (
Aval) was determined in accordance with PN-ISO 660:2009 standard and calculated by using Equation (3)
where
Vb and
Vs are volumes in mL of potassium hydroxide used for the blank and sample, respectively;
CKOH is the concentration of potassium hydroxide solution in mol/dm
3;
ms is the mass of the sample in g. Samples of approximately 10 g were used.
Epoxy value (
Eval) was determined in accordance with the PN-87/C-89085/13 standard. The method involves a quantitative reaction of hydrochloric acid with epoxy group at room temperature and titration of the hydrochloric acid excess using a solution of sodium hydroxide in methanol in the presence of cresol red as an indicator. Eval was calculated by using Equation (4)
where
Vb and
Vs are volumes in ml of sodium hydroxide used for the blank and sample, respectively;
CNaOH is the concentration of potassium hydroxide solution in mol/dm
3;
ms is the mass of the sample in g. Samples of approximately 0.3 g were used.
Viscosity was determined using a Haake Viscotester 7 plus from Thermo Fisher Scientific (Waltham, MA, USA) at 25 °C (298 K).
Water content (%H2O) was determined by Karl Fisher method in accordance with PN-81/C-04959 with the use of a TitroLine TA 05 plus device manufactured by SCHOTT Instruments GmbH (Meinz, Germany).
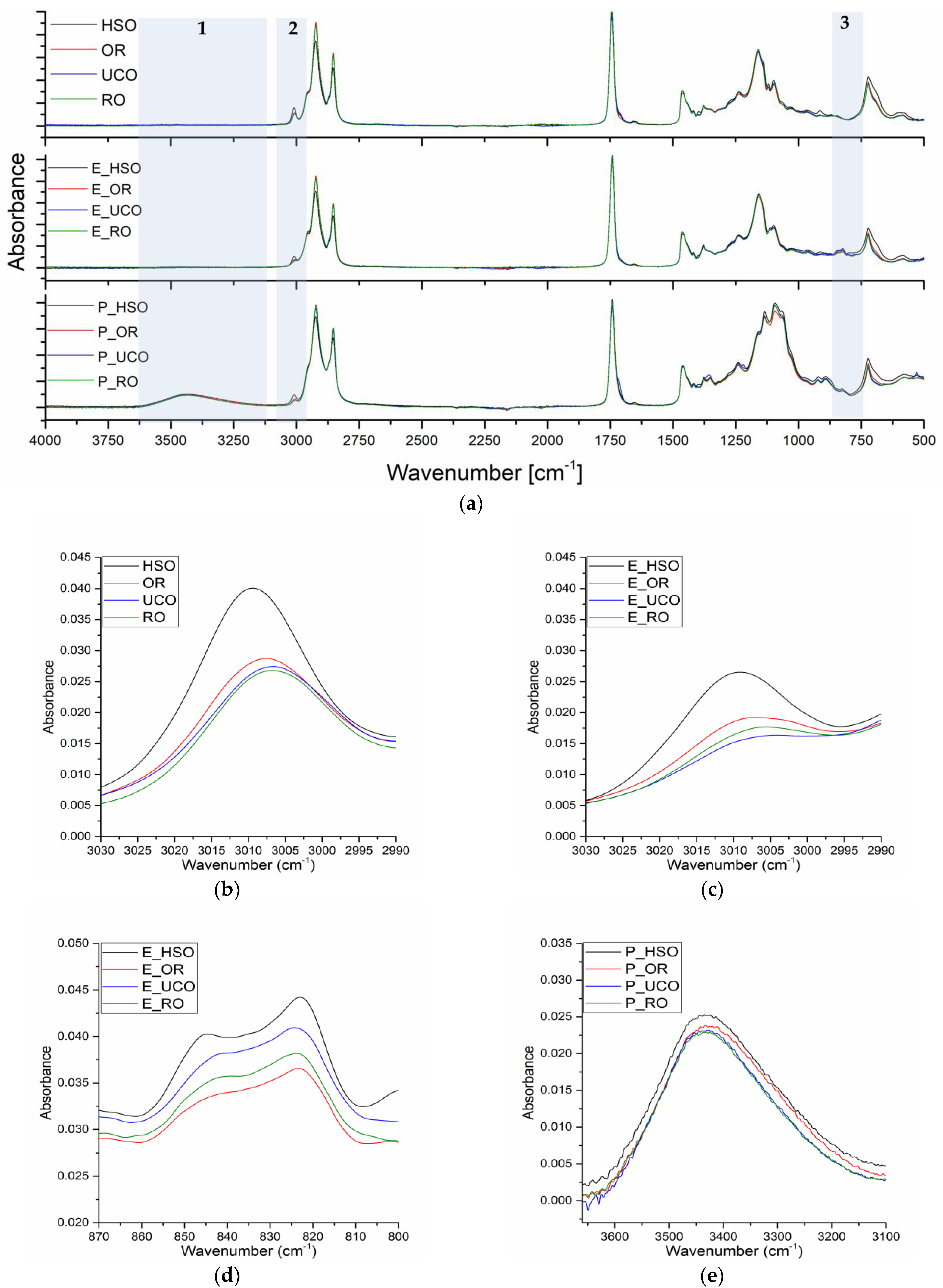
The chemical structures were analysed using an FTIR spectrometer model Nicolet iS5 from Thermo Fisher Scientific (Waltham, MA, USA), equipped with an ATR (attenuated total reflection) accessory with a diamond crystal in the infrared range of 4000–500 cm−1.
The number average molecular weight (Mn) and weight average molecular weight (Mw) were determined using gel permeation chromatography (GPC) analysis with the use of a chromatograph equipped with a refractometric detector. The analyses were performed at 25 °C. Tetrahydrofuran was used as an eluent and its flow rate was fixed at 1 mL/min. The functionality (f) refers to the number of OH groups per molecule was calculated following Formula (5):
2.4. Characterization of PUR Foams
The apparent density of foam samples was calculated in accordance with the ISO 845:2006 standard as a ratio of the masses and volumes of samples.
Thermal conductivity coefficient was measured with the use of a heat flow meter instrument Fox200 (TA Instruments, DE, USA) in accordance with the ISO 8301 standard. The foam samples with dimensions of 5 cm × 20 cm × 20 cm were placed in the apparatus. Heat flow was measured at plate temperatures of 0 and 20 °C.
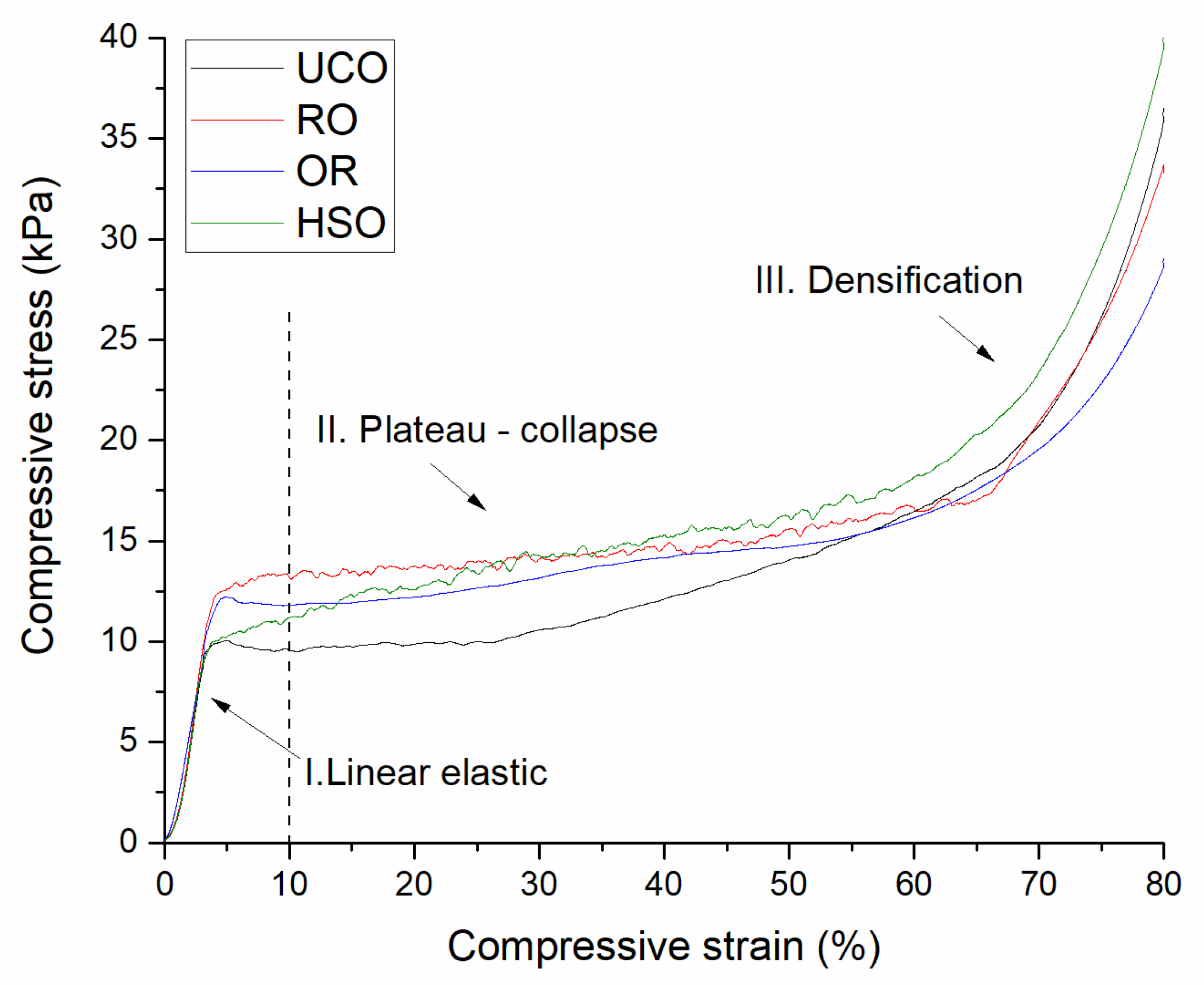
Compressive strength was measured at 10% deformation using a Zwick Roell testing machine (Zwick Roell Group, Ulm, Germany) according to the PN-EN 826:2013-07 standard. The mechanical properties of the foams were evaluated in a direction perpendicular to the foam growth direction. Eight cylindrical-shaped samples with a diameter of 40 mm and a height of 40 mm cut from the cores of two polyurethane foams were tested.
The short-term water absorption test was carried out according to the PN-EN 1609:2013 standard.
Water vapour permeability (δ) and water vapour diffusion resistance factor (μ) were determined according to the PN-EN 12086:2013 standard. The following conditions were used in the test: 23 °C, 85% relative humidity, anhydrous calcium chloride was used as a desiccant. Six samples from each foam were tested. The weight of the test assembly was measured at regular 24 h intervals until the change in mass was constant within ± 5% of the mean value.
The foam morphology was examined using a scanning electron microscope (SEM) Apreo 2 S LoVac (Thermo Fisher Scientific, Waltham, MA, USA) configured with a secondary electron detector in low vacuum mode and an acceleration voltage of 22 kV. The samples were coated with a 4 nm layer of carbon. Magnifications of 70 and 250 times were used. The analysis of the foam morphology (cell cross-section area) was performed using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA,
https://imagej.nih.gov/ij/, accessed on 12 December 2022, ver. 1.53 t)
2.5. Synthesis of Epoxidized Vegetable Oils
The reaction setup consisted of a water bath in which four 1 dm
3 round-bottom flasks equipped with identical stirrers set at 500 rpm, a thermometer and a reflux condenser were placed. Amounts of 250 g of oil and 37.5 g of Amberlite IR 120 H catalyst (15% by weight of oil) were added to the flasks and the bath temperature was set to 65 °C. After the set temperature was reached, 15 g of GAA and 133 g of HP were added. The amount of GAA and HP were calculated according to Equations (6) and (7), assuming that regardless of the oil used and its initial Ival, the final Eval was expected to be 0.2 mol/100 g.
where
mGAA is the mass of glacial acetic acid [g],
mHP is the mass of hydrogen peroxide [g],
EVexp is the expected
Ev value [mol/100 g],
moil is the mass of the oil subjected to epoxidation [g].
The amounts of GAA and HP were 0.5 and 2 moles, respectively, for every 1 mol of unsaturated bonds subjected to the epoxidation reaction. The value of Eval of the epoxidized oils was selected based on previous studies [
30], which showed that Eval above 0.2 mol/100 g led to a significant increase in the viscosity of the bio-polyol with a slight increase in the hydroxyl group content. In addition, reducing the use of acetic acid and hydrogen peroxide is beneficial from the ecological and economic point of view. The course of the reaction was monitored by periodically (every 20 min) measuring Eval. The effect of the chemical composition of the oils on the epoxidation process was studied by running the reactions for 9 h. Then, the epoxidation reactions of unrefined oils were carried out for 3 h and, in the case of UCO, for 4 h 20 min. After the reaction was completed, the oils were washed 4 times with 500 mL of warm water and distilled under reduced pressure.
2.6. Synthesis of Bio-Polyols
The syntheses of bio-polyols were carried out in a 0.5 dm3 reactor equipped with a heating mantle, a mechanical stirrer and a thermometer. Each time, 200 g of epoxidized oil was used. After the temperature of epoxidized oil reached 80 °C, a mixture of DEG (in stoichiometric amount to epoxy group content) and 0.4 g of tetrafluoroboric acid (0.2% by weight of the epoxidized oil) was added to the reactor while maintaining intensive mixing. The change in the epoxy value was monitored by taking samples every 10 min. The reaction was carried out until all the epoxy groups were reacted (titrated Eval = 0 mol/100 g).
2.7. Preparation of Polyurethane Foams
The PUR foams were obtained using a one-step two-component method. One of the components (A) consisted of bio-polyol, surfactants, catalysts, water and a flame retardant. The second component (B) consisted of isocyanate. The volume ratio of components was 1:1. The temperature of components A and B was 40 °C. Component A was mixed for 30 s with a mechanical stirrer and then component B was added and stirred for 3 s. The mixture was poured into a horizontal mould that provided free growth of the foam. The use of a mechanical stirrer affects the properties of foams but gives basic information on whether a given polyurethane system is suitable for industrial-scale testing with high-pressure spray machines. Samples were cut out 24 h after obtaining the foams. The formulation of the polyurethane systems is shown in
Table 2.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}