
Smart Materials Prediction: Applying Machine Learning to Lithium Solid-State Electrolyte





Abstract
:1. Introduction

2. Data Sets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Website | Overview |
|---|---|---|
| ICSD | fiz-karlsruhe.de/icsd | Provides information on the crystal structures of all inorganic compounds without C-H bonds, except for metals and alloys [30] |
| Material project | materialsproject.org | Uses high-throughput computing to uncover the properties of all known inorganic materials [28] |
| AFLOW | aflowlib.org | The library is mainly composed of chalcogenide data; users can download the whole database [31] |
| OQMD | oqmd.org | The library is mainly composed of chalcogenide data; users can download the whole database [32] |
| Computational Materials Repository | cmr.fysik.dtu.dk | Supports the collection, storage, retrieval, analysis and sharing of data produced by many electronic-structure simulators [33] |
| Crystallography Open Database | crystallography.net | Provides capabilities for all registered users to deposit published and so far unpublished structures as personal communications or pre-publication depositions. Such a setup simultaneously enables the COD database extension by many users [34] |
| MATGEN | matgen.nscc-gz.cn | Contains crystal structure information, ion migration channel connectivity information and 3D channel maps for over 29,000 inorganic compounds [29] |

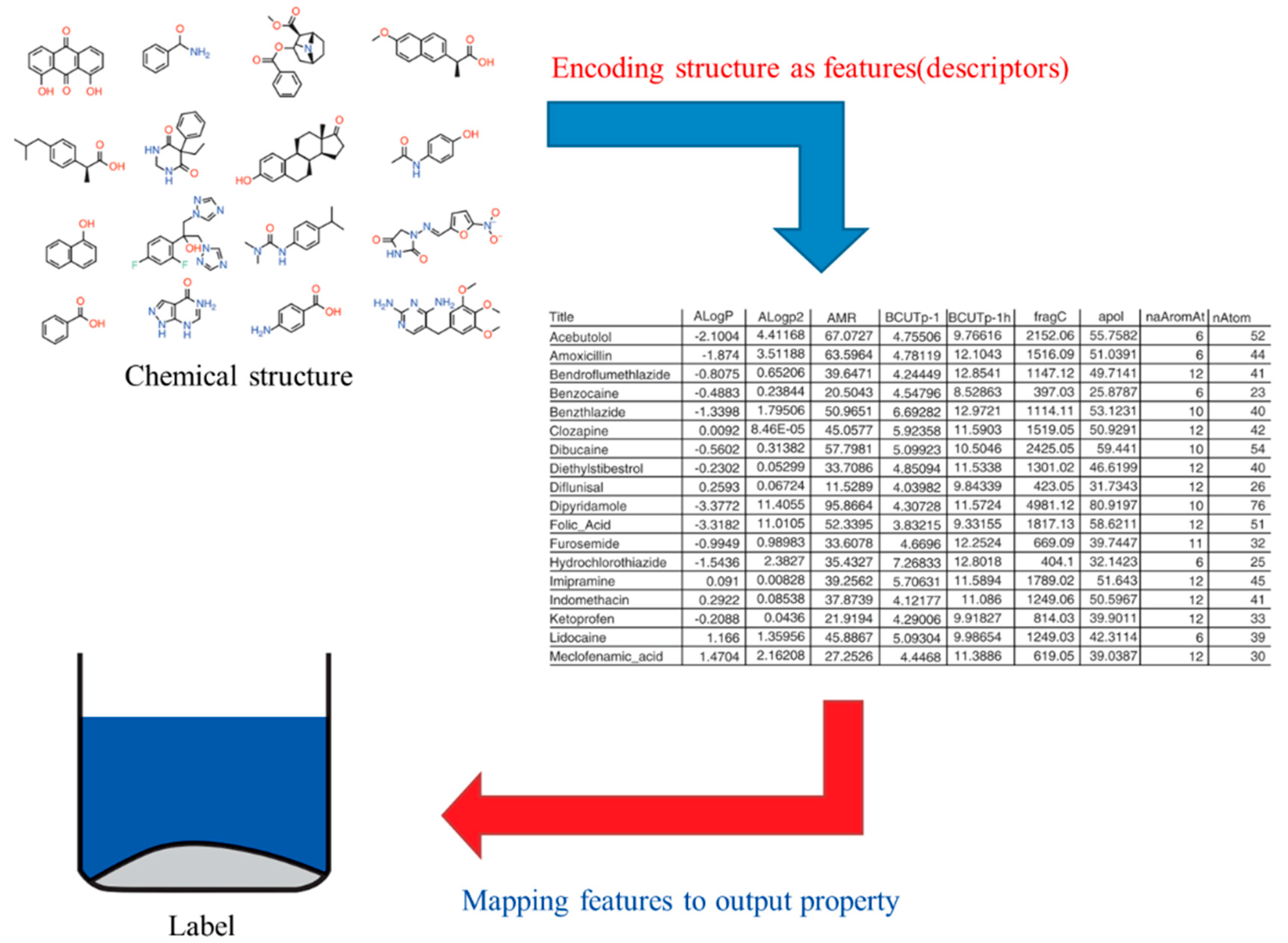
3. Descriptor
4. Construction of ML Model
4.1. Supervised Learning Model
4.2. Unsupervised Learning Model
4.3. Semi-Supervised Learning Model
5. Algorithm Application
6. Algorithm Optimization
7. Views and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ceder, G. Computational materials science-predicting properties from scratch. Science 1998, 280, 1099–1100. [Google Scholar] [CrossRef]
- Bleidorn, W.; Hopwood, C.J. Using machine learning to advance personality assessment and theory. Pers. Soc. Psychol. Rev. 2019, 23, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Karpatne, A.; Ebert-Uphoff, I.; Ravela, S.; Babaie, H.A.; Kumar, V. Machine learning for the geosciences: Challenges and opportunities. IEEE Trans. Knowl. Data Eng. 2019, 31, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Phung, D.; Tran, T.; Gupta, S.; Rana, S.; Karmakar, C.; Shilton, A.; Yearwood, J.; Dimitrova, N.; Ho, T.B.; et al. Guidelines for developing and reporting machine learning predictive models in biomedical research: A multidisciplinary view. J. Med Internet Res. 2016, 18, e323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, N.; Mao, B.; Tang, F.; Kawamoto, Y.; Liu, J. Ten challenges in advancing machine learning technologies toward 6G. IEEE Wirel. Commun. 2020, 27, 96–103. [Google Scholar] [CrossRef]
- Agrawal, A.; Choudhary, A. Perspective: Materials informatics and big data: Realization of the “fourth paradigm” of science in materials science. APL Mater. 2016, 4, 053208. [Google Scholar] [CrossRef] [Green Version]
- Sendek, A.D.; Cubuk, E.D.; Antoniuk, E.R.; Cheon, G.; Cui, Y.; Reed, E.J. Machine learning-assisted discovery of solid Li-Ion conducting materials. Chem. Mater. 2019, 31, 342–352. [Google Scholar] [CrossRef]
- Wang, Z.; Lin, X.; Han, Y.; Cai, J.; Wu, S.; Yu, X.; Li, J. Harnessing artificial intelligence to holistic design and identification for solid electrolytes. Nano Energy 2021, 89, 106337. [Google Scholar] [CrossRef]
- Himanen, L.; Geurts, A.; Foster, A.S.; Rinke, P. Data-driven materials science: Status, challenges, and perspectives. Adv. Sci. 2019, 6, 1900808. [Google Scholar] [CrossRef]
- Fan, E.; Li, L.; Wang, Z.; Lin, J.; Huang, Y.; Yao, Y.; Chen, R.; Wu, F. Sustainable recycling technology for Li-Ion batteries and beyond: Challenges and future prospects. Chem. Rev. 2020, 120, 7020–7063. [Google Scholar] [CrossRef] [PubMed]
- Mcdowell, M.T.; Cortes, F.J.Q.; Thenuwara, A.C.; Lewis, J.A. Toward high-capacity battery anode materials: Chemistry and mechanics intertwined. Chem. Rev. 2020, 32, 8755–8771. [Google Scholar] [CrossRef]
- Banerjee, A.; Wang, X.; Fang, C.; Wu, E.A.; Meng, Y.S. Interfaces and interphases in all-solid-state batteries with inorganic solid electrolytes. Chem. Rev. 2020, 120, 6878–6933. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, Q.; Yu, X.; Chen, L.; Li, H. Approaching practically accessible solid-state batteries: Stability issues related to solid electrolytes and interfaces. Chem. Rev. 2020, 120, 6820–6877. [Google Scholar] [CrossRef] [PubMed]
- Huggins, R.A. Recent results on lithium ion conductors. Electrochim. Acta 1977, 22, 773–781. [Google Scholar] [CrossRef]
- Brissot, C.; Rosso, M.; Chazalviel, J.-N.; Lascaud, S. Dendritic growth mechanisms in lithium/polymer cells. J. Power Sources 1999, 81-82, 925–929. [Google Scholar] [CrossRef]
- Kamaya, N.; Homma, K.; Yamakawa, Y.; Hirayama, M.; Kanno, R.; Yonemura, M.; Kamiyama, T.; Kato, Y.; Hama, S.; Kawamoto, K.; et al. A Lithium Superionic Conductor. Nat. Mater. 2011, 10, 682–686. [Google Scholar] [CrossRef]
- Kerman, K.; Luntz, A.; Viswanathan, V.; Chiang, Y.-M.; Chen, Z. Review—Practical challenges hindering the development of solid state Li Ion batteries. J. Electrochem. Soc. 2017, 164, A1731–A1744. [Google Scholar] [CrossRef]
- Rajan, K. Materials Informatics. Mater. Today 2005, 8, 38–45. [Google Scholar] [CrossRef]
- Kaufman, L.; Ågren, J. CALPHAD, first and second generation—Birth of the materials genome. Scr. Mater. 2014, 70, 3–6. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Gonze, X.; Beuken, J.-M.; Caracas, R.; Detraux, F.; Fuchs, M.; Rignanese, G.-M.; Sindic, L.; Verstraete, M.; Zerah, G.; Jollet, F.; et al. First-principles computation of material properties: The ABINIT software project. Comput. Mater. Sci. 2002, 25, 478–492. [Google Scholar] [CrossRef]
- Greeley, J.; Nørskov, J.K. Large-scale, density functional theory-based screening of alloys for hydrogen evolution. Surf. Sci. 2007, 601, 1590–1598. [Google Scholar] [CrossRef]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Gonze, X.; Amadon, B.; Anglade, P.M.; Beuken, J.-M.; Bottin, F.; Boulanger, P.; Bruneval, F.; Caliste, D.; Caracas, R.; Côté, M.; et al. ABINIT: First-principles approach to material and nanosystem properties. Comput. Phys. Commun. 2009, 180, 2582–2615. [Google Scholar] [CrossRef]
- Ong, S.P.; Richards, W.D.; Jain, A.; Hautier, G.; Kocher, M.; Cholia, S.; Gunter, D.; Chevrier, V.L.; Persson, K.A.; Ceder, G. Python materials genomics (pymatgen): A robust, open-source python library for materials analysis. Comput. Mater. Sci. 2013, 68, 314–319. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.H.; Rose, F.; Toher, C.; Levy, O.; Yang, K.; Buongiorno Nardelli, M.; Curtarolo, S. A RESTful API for Exchanging Materials Data in the AFLOWLIB.org Consortium. Comput. Mater. Sci. 2014, 93, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The materials project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; He, B.; Zhao, Q.; Zou, Z.; Chi, S.; Mi, P.; Ye, A.; Li, Y.; Wang, D.; Avdeev, M.; et al. A database of ionic transport characteristics for over 29,000 inorganic compounds. Adv. Funct. Mater. 2020, 30, 2003087. [Google Scholar] [CrossRef]
- Bergerhoff, G.; Hundt, R.; Sievers, R.; Brown, I.D. The inorganic crystal structure data base. J. Chem. Inf. Comput. Sci. 1983, 23, 66–69. [Google Scholar] [CrossRef]
- Curtarolo, S.; Setyawan, W.; Wang, S.; Xue, J.; Yang, K.; Taylor, R.H.; Nelson, L.J.; Hart, G.L.W.; Sanvito, S.; Buongiorno-Nardelli, M.; et al. AFLOWLIB.ORG: A distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 2012, 58, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Saal, J.E.; Kirklin, S.; Aykol, M.; Meredig, B.; Wolverton, C. Materials design and discovery with high-throughput density functional theory: The open quantum materials database (OQMD). JOM 2013, 65, 1501–1509. [Google Scholar] [CrossRef]
- Landis, D.D.; Hummelshoj, J.S.; Nestorov, S.; Greeley, J.; Dulak, M.; Bligaard, T.; Norskov, J.K.; Jacobsen, K.W. The computational materials repository. Comput. Sci. Eng. 2012, 14, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Gražulis, S.; Daškevič, A.; Merkys, A.; Chateigner, D.; Lutterotti, L.; Quirós, M.; Serebryanaya, N.R.; Moeck, P.; Downs, R.T.; Le Bail, A. Crystallography open database (COD): An open-access collection of crystal structures and platform for world-wide collaboration. Nucleic Acids Res. 2012, 40, D420–D427. [Google Scholar] [CrossRef] [PubMed]
- Sendek, A.D.; Yang, Q.; Cubuk, E.D.; Duerloo, K.-A.N.; Cui, Y.; Reed, E.J. Holistic computational structure screening of more than 12,000 candidates for solid Lithium-Ion conductor materials. Energy Environ. Sci. 2017, 10, 306–320. [Google Scholar] [CrossRef]
- Mahbub, R.; Huang, K.; Jensen, Z.; Hood, Z.D.; Rupp, J.L.M.; Olivetti, E.A. Text mining for processing conditions of solid-state battery electrolytes. Electrochem. Commun. 2020, 121, 106860. [Google Scholar] [CrossRef]
- Jensen, Z.; Kim, E.; Kwon, S.; Gani, T.Z.H.; Román-Leshkov, Y.; Moliner, M.; Corma, A.; Olivetti, E. A machine learning approach to zeolite synthesis enabled by automatic literature data extraction. ACS Cent. Sci. 2019, 5, 892–899. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, L.M.; Vybiral, J.; Levchenko, S.V.; Draxl, C.; Scheffler, M. Big data of materials science: Critical role of the descriptor. Phys. Rev. Lett. 2015, 114. [Google Scholar] [CrossRef] [Green Version]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef]
- Ramprasad, R.; Batra, R.; Pilania, G.; Mannodi-Kanakkithodi, A.; Kim, C. Machine learning in materials informatics: Recent applications and prospects. NPJ Comput. Mater. 2017, 3, 3. [Google Scholar] [CrossRef]
- Von Lilienfeld, O.A.; Ramakrishnan, R.; Rupp, M.; Knoll, A. Fourier series of atomic radial distribution functions: A molecular fingerprint for machine learning models of quantum chemical properties. Int. J. Quantum Chem. 2015, 115, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Bartók, A.P.; Kondor, R.; Csányi, G. On representing chemical environments. Phys Rev. B 2013, 87, 219902. [Google Scholar] [CrossRef]
- Isayev, O.; Oses, C.; Toher, C.; Gossett, E.; Curtarolo, S.; Tropsha, A. Universal fragment descriptors for predicting properties of inorganic crystals. Nat. Commun. 2017, 8, 15679. [Google Scholar] [CrossRef] [PubMed]
- Rupp, M.; Tkatchenko, A.; Müller, K.-R.; Von Lilienfeld, O.A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012, 108. [Google Scholar] [CrossRef]
- Jäger, M.O.J.; Morooka, E.V.; Federici, C.F.; Himanen, L.; Foster, A.S. Machine learning hydrogen adsorption on nanoclusters through structural descriptors. PJ Comput. Mater. 2018, 4, 37. [Google Scholar] [CrossRef]
- Li, Y.; Yu, J.H. New stories of zeolite structures: Their descriptions, determinations, predictions, and evaluations. Chem. Rev. 2014, 114, 7268–7316. [Google Scholar] [CrossRef]
- Faber, F.A.; Hutchison, L.; Huang, B.; Gilmer, J.; Schoenholz, S.S.; Dahl, G.E.; Vinyals, O.; Kearnes, S.; Riley, P.F.; Von Lilienfeld, O.A. Prediction errors of molecular machine learning models lower than hybrid DFT error. J. Chem. Theory Comput. 2017, 13, 5255–5264. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Choi, E.; Kim, M.; Min, K. Machine learning-aided materials design platform for predicting the mechanical properties of Na-Ion solid-state electrolytes. ACS Appl. Energy Mater. 2021, 4, 7862–7869. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Chen, D.; Jiang, Y.; Nie, Z.; Pan, F. Encoding the atomic structure for machine learning in materials science. WIREs Comput. Mol. Sci. 2022, 12, e1558. [Google Scholar] [CrossRef]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom pairs as molecular features in structure-activity studies: Definition and applications. J. Chem. Inf. Comput. Sci. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Z.Q.; Su, J.; Li, J.F. Data mining new energy materials from structure databases. Renew. Sustain. Energy Rev. 2019, 107, 554–567. [Google Scholar] [CrossRef]
- Adnan, S.B.R.S.; Mohamed, N.S. Electrical properties of novel Li4.08Zn0.04Si0.96O4 ceramic electrolyte at high temperatures. Ionics 2014, 20, 1641–1650. [Google Scholar] [CrossRef]
- Zhao, W.; Yi, J.; He, P.; Zhou, H. Solid-state electrolytes for Lithium-Ion batteries: Fundamentals, challenges and perspectives. Electrochem. Energy Rev. 2019, 2, 574–605. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Goto, M.; Kato, R.; Tanaka, Y.; Kagawa, Y. Thermal conductivity of ZnO thin film produced by reactive sputtering. J. Appl. Phys. 2012, 111, 084320. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Tanaka, T.; Komori, T.; Fujii, M.; Mizuno, H.; Itoh, S.; Takada, T.; Fujita, E.; Xu, Y. Essential structural and experimental descriptors for bulk and grain boundary conductivities of li solid electrolytes. Sci. Technol. Adv. Mater. 2020, 21, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.O. Machine learning methods in chemoinformatics. WIREs Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, K.; Seko, A.; Koyama, Y.; Kuwabara, A.; Kishida, I.; Shitara, K.; Fisher, C.A.J.; Moriwake, H.; Tanaka, I. Accelerated materials design of lithium superionic conductors based on first-principles calculations and machine learning algorithms. Adv. Energy Mater. 2013, 3, 980–985. [Google Scholar] [CrossRef]
- Lazarovits, J.; Sindhwani, S.; Tavares, A.J.; Zhang, Y.; Song, F.; Audet, J.; Krieger, J.R.; Syed, A.M.; Stordy, B.; Chan, W.C.W. Supervised learning and mass spectrometry predicts the in vivo fate of nanomaterials. ACS Nano 2019, 13, 8023–8034. [Google Scholar] [CrossRef]
- Timoshenko, J.; Wrasman, C.J.; Luneau, M.; Shirman, T.; Cargnello, M.; Bare, S.R.; Aizenberg, J.; Friend, C.M.; Frenkel, A.I. Probing atomic distributions in mono- and bimetallic nanoparticles by supervised machine learning. Nano Lett. 2019, 19, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.Y.; Bousquet, O.; Lal, T.N.; Weston, J.; Scholkopf, B. Learning with Local and Global Consistency. In Advances in Neural Information Processing Systems; The MIT Press: Cambridge, UK, 2004; Volume 16, pp. 321–328. [Google Scholar]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Xie, T.; Maheshwari, C.; Grossman, J.C.; Viswanathan, V. Machine learning enabled computational screening of inorganic solid electrolytes for suppression of dendrite formation in lithium metal anodes. ACS Cent. Sci. 2018, 4, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubuk, E.D.; Sendek, A.D.; Reed, E.J. Screening billions of candidates for solid Lithium-Ion conductors: A transfer learning approach for small data. J. Chem. Phys. 2019, 150, 214701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajibabaei, A.; Kim, K.S. Universal machine learning interatomic potentials: Surveying solid electrolytes. J. Phys. Chem. Lett. 2021, 12, 8115–8120. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.J.; Kuncheva, L.I.; Alonso, C.J. Rotation forest: A new classifier ensemble method. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1619–1630. [Google Scholar] [CrossRef]
- Choi, E.; Jo, J.; Kim, W.; Min, K. Searching for mechanically superior solid-state electrolytes in Li-Ion batteries via data-driven approaches. ACS Trans. Pattern Anal. Mach. Intell. 2021, 13, 42590–42597. [Google Scholar] [CrossRef]
- Sendek, A.D.; Cheon, G.; Pasta, M.; Reed, E.J. Quantifying the search for solid li-ion electrolyte materials by Anion: A data-driven perspective. J. Phys. Chem. C 2020, 124, 8067–8079. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Zhang, X.; Zhou, Z. Machine learning: Accelerating materials development for energy storage and conversion. InfoMat 2020, 2, 553–576. [Google Scholar] [CrossRef]
- Gu, J.; Wang, Z.; Kuen, J.; Ma, L.; Shahroudy, A.; Shuai, B.; Liu, T.; Wang, X.; Wang, G.; Cai, J.; et al. Recent advances in convolutional neural networks. Pattern Recognit. 2018, 77, 354–377. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Grossman, J.C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 2018, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; He, X.; Chen, Z.; Bai, Q.; Nolan, A.M.; Roberts, C.A.; Banerjee, D.; Matsunaga, T.; Mo, Y.; Ling, C. Unsupervised discovery of solid-state Lithium Ion conductors. Nat. Commun. 2019, 10, 5260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Hand, D.J.; Yu, K. Idiot’s bayes? Not so stupid after all? Int. Stat. Rev. 2001, 69, 385–398. [Google Scholar]
- Weher, E.; Edwards, A.L. An Introduction to Linear Regression and Correlation; A Series of Books in Psychology; W. H. Freeman and Comp.: San Francisco, CA, USA, 1976; 213p. [Google Scholar]
- Friedman, J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001, 29, 1189–1232. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Duquesnoy, M.; Tan, D.H.S.; Doux, J.-M.; Yang, H.; Deysher, G.; Ridley, P.; Franco, A.A.; Meng, Y.S.; Chen, Z. Fabrication of high-quality thin solid-state electrolyte films assisted by machine learning. ACS Energy Lett. 2021, 1639–1648. [Google Scholar] [CrossRef]
- Meredig, B.; Antono, E.; Church, C.; Hutchinson, M.; Ling, J.; Paradiso, S.; Blaiszik, B.; Foster, I.; Gibbons, B.; Hattrick-Simpers, J.; et al. Can machine learning identify the next high-temperature superconductor? Examining extrapolation performance for materials discovery. Mol. Syst. Des. Eng. 2018, 3, 819–825. [Google Scholar] [CrossRef] [Green Version]
- Kononova, O.; He, T.J.; Huo, H.Y.; Trewartha, A.; Olivetti, E.A.; Ceder, G. Opportunities and challenges of text mining in materials research. iScience 2021, 24, 102155. [Google Scholar] [CrossRef]
- Pu, J.; Shao, H.; Gao, B.; Zhu, Z.; Zhu, Y.; Rao, Y.; Xiang, Y. Matexplorer: Visual exploration on predicting ionic conductivity for solid-state electrolytes. IEEE Rans. Vis. Comput. Graph. 2022, 28, 65–75. [Google Scholar] [CrossRef]
- Raccuglia, P.; Elbert, K.C.; Adler, P.D.F.; Falk, C.; Wenny, M.B.; Mollo, A.; Zeller, M.; Friedler, S.A.; Schrier, J.; Norquist, A.J. Machine-learning-assisted materials discovery using failed experiments. Nature 2016, 533, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Sung, F.; Zhang, L.; Xiang, T.; Hospedales, T.; Yang, Y. Learning to learn: Meta-critic networks for sample efficient learning. arXiv 2017, arXiv:1706.09529. [Google Scholar]
- Artrith, N.; Butler, K.T.; Coudert, F.-X.; Han, S.; Isayev, O.; Jain, A.; Walsh, A. Best practices in machine learning for chemistry. Nat. Chem. 2021, 13, 505–508. [Google Scholar] [CrossRef] [PubMed]


| Descriptor | Overview |
|---|---|
| Coulomb matrix (CM) | It represents an atom-by-atom square matrix. The structure is encoded according to the Coulomb force between each pair of atomic charges, in which the off-diagonal element is the Coulomb nuclear repulsion term between atomic pairs [44]. |
| Smooth overlap of atomic positions (SOAP) | SOAP is a translation, rotation and arrangement-invariant descriptor for obtaining the translation, rotation and arrangement of atomic groups, which is the basis for developing various ML interatomic potentials [42]. |
| Diffraction fingerprint | The diffraction fingerprint emphasizes the global characteristics of infinite periodic crystals, which are excited by the properties of the Fourier transform [49]. |
| Topological descriptor | Commonly referred to as path-based fingerprints, chemical structures are encoded according to combinations of atom types and paths between them (e.g., atom-pair fingerprints). They are essentially graph-based descriptors [50]. |
| Quantum descriptors | Based on first-principles calculations. The descriptors calculated from the wave function include energy levels, dipole moments, polarizability, etc. The quantum descriptors are often considered to be more versatile since they better represent the properties, but more difficult and time-consuming to obtain than the other descriptors for the structure [51]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Q.; Chen, K.; Liu, F.; Zhao, M.; Liang, F.; Xue, D. Smart Materials Prediction: Applying Machine Learning to Lithium Solid-State Electrolyte. Materials 2022, 15, 1157. https://doi.org/10.3390/ma15031157
Hu Q, Chen K, Liu F, Zhao M, Liang F, Xue D. Smart Materials Prediction: Applying Machine Learning to Lithium Solid-State Electrolyte. Materials. 2022; 15(3):1157. https://doi.org/10.3390/ma15031157
Chicago/Turabian StyleHu, Qianyu, Kunfeng Chen, Fei Liu, Mengying Zhao, Feng Liang, and Dongfeng Xue. 2022. "Smart Materials Prediction: Applying Machine Learning to Lithium Solid-State Electrolyte" Materials 15, no. 3: 1157. https://doi.org/10.3390/ma15031157
APA StyleHu, Q., Chen, K., Liu, F., Zhao, M., Liang, F., & Xue, D. (2022). Smart Materials Prediction: Applying Machine Learning to Lithium Solid-State Electrolyte. Materials, 15(3), 1157. https://doi.org/10.3390/ma15031157