
Application of DFT Simulation to the Investigation of Hydrogen Embrittlement Mechanism and Design of High Strength Low Alloy Steel

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Experiments
2.2. DFT Calculations
3. Results and Discussion
4. Conclusions
- (a)
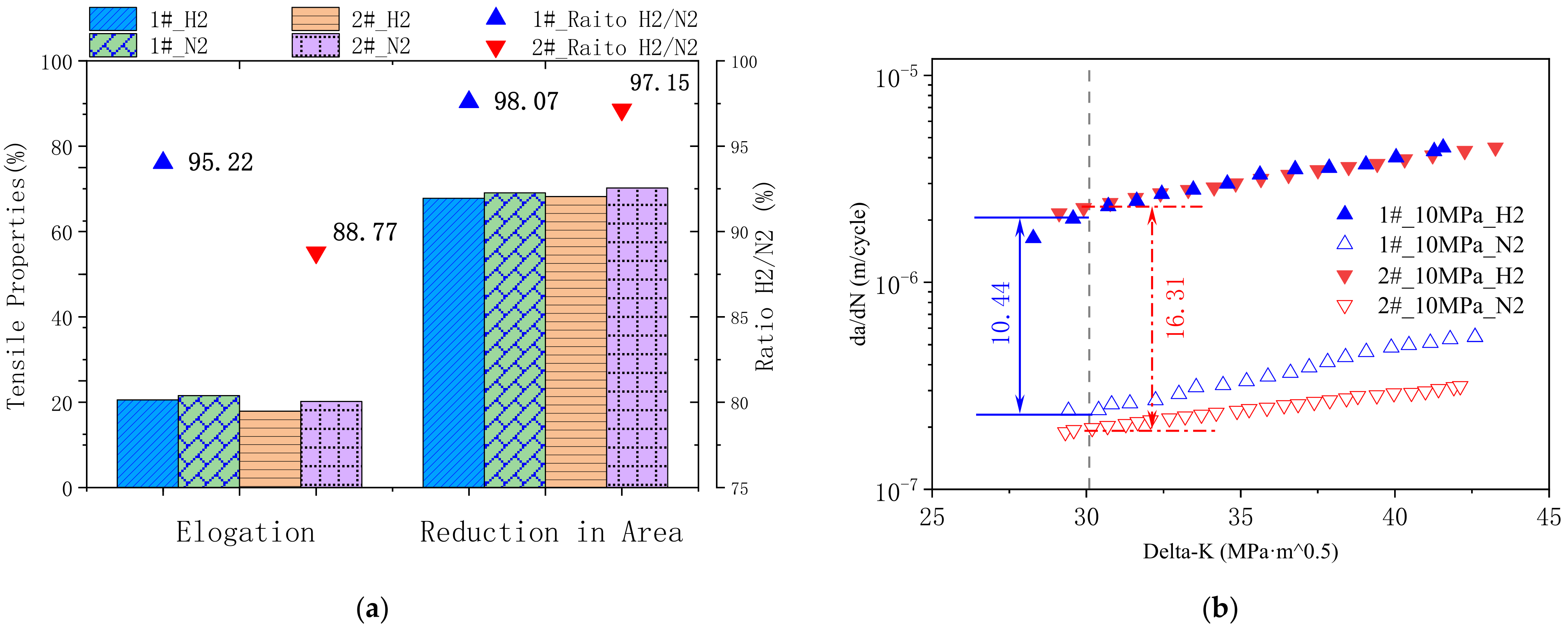
- Model steel 1# with higher content of Ni, Cr, and Mo behaves with lower susceptibility of hydrogen embrittlement in high-pressure hydrogen gas;
- (b)
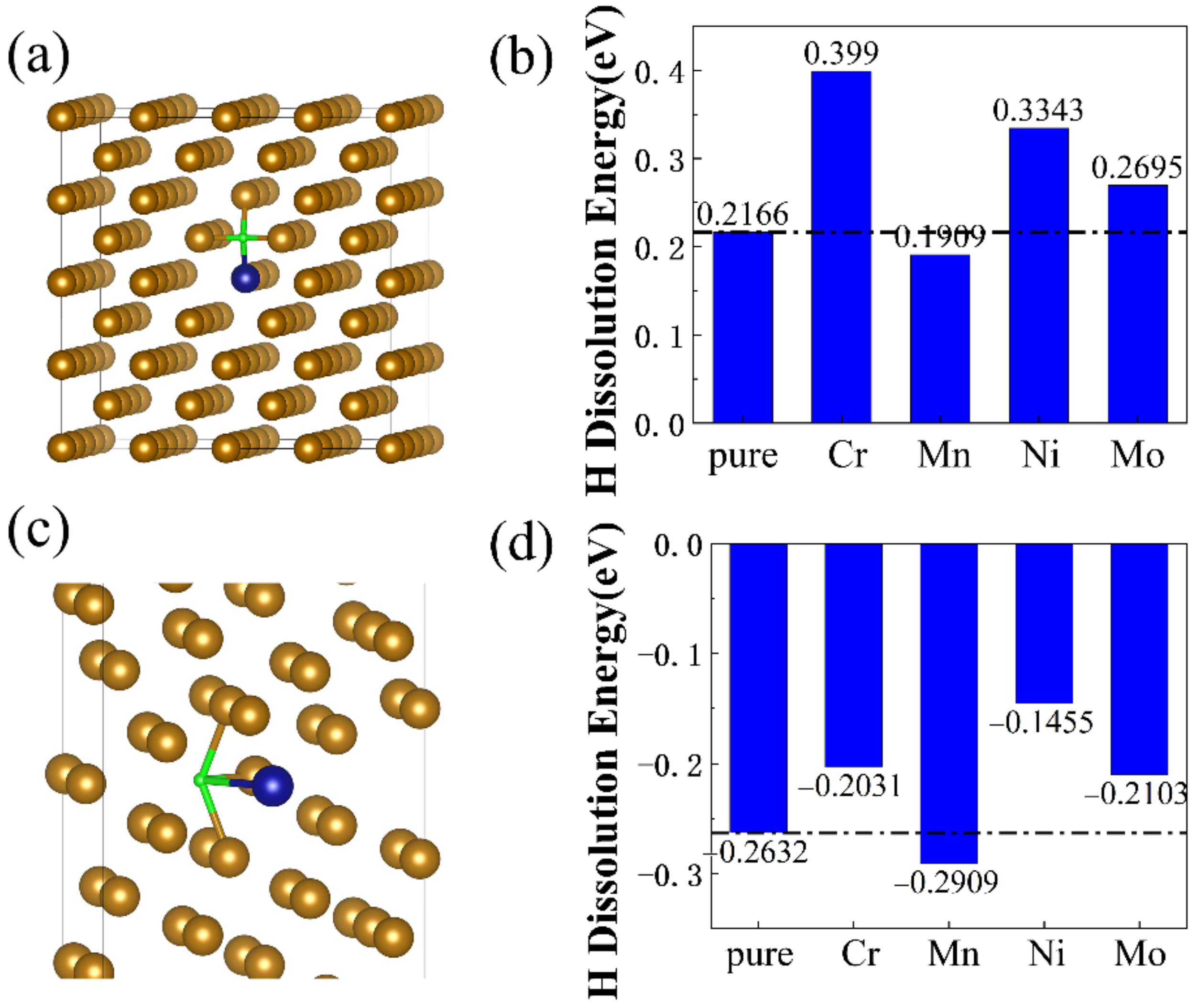
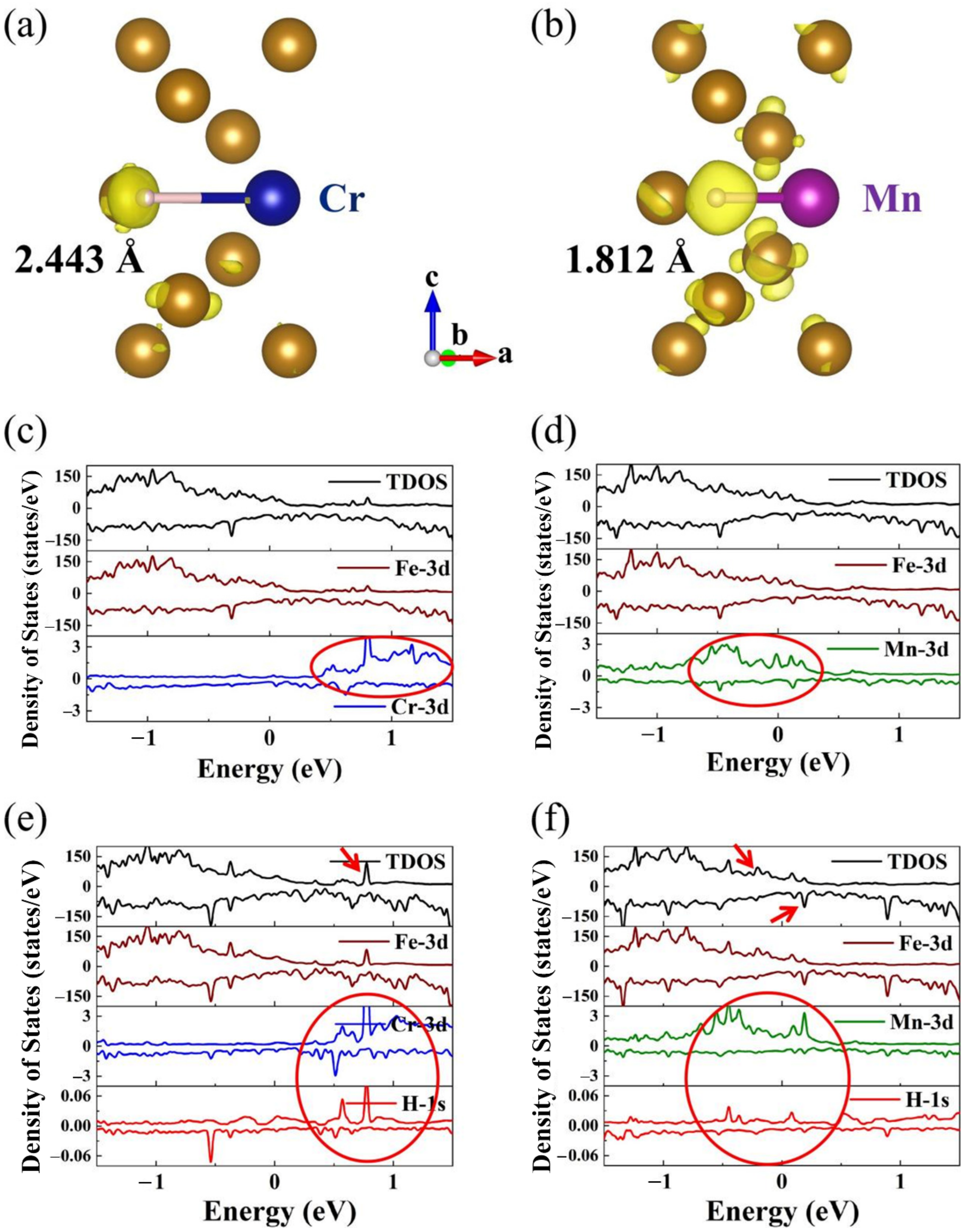
- Alloying elements Cr, Ni, and Mo doping in bulk and Σ5 GB structures could increase H dissolution energies, while Mn doping could decrease H dissolution energies;
- (c)
- In comparison to bulk structures, GB structures show a higher diffusion barrier of H atoms and a lower H diffusion coefficient at grain boundaries;
- (d)
- Alloying elements significantly affect the hydrogen diffusion behavior, especially at Σ5 GB. The hydrogen diffusion coefficient for Ni dopingΣ5 GB structures increases by approximately six orders of magnitude.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andersson, J.; Grönkvist, S. Large-scale storage of hydrogen. Int. J. Hydrog. Energy 2019, 44, 11901–11919. [Google Scholar] [CrossRef]
- Liu, M.; Wang, J.; Ke, W.; Han, E.-H. Corrosion behavior of X52 anti-H2S pipeline steel exposed to high H2S concentration solutions at 90 C. J. Mater. Sci. Technol. 2014, 30, 504–510. [Google Scholar] [CrossRef]
- Xue, H.; Cheng, Y. Characterization of inclusions of X80 pipeline steel and its correlation with hydrogen-induced cracking. Corros. Sci. 2011, 53, 1201–1208. [Google Scholar] [CrossRef]
- Sun, B.; Wang, D.; Lu, X.; Wan, D.; Ponge, D.; Zhang, X. Current Challenges and Opportunities Toward Understanding Hydrogen Embrittlement Mechanisms in Advanced High-Strength Steels: A Review. Acta Metall. Sin. 2021, 34, 741–754. [Google Scholar] [CrossRef]
- Robertson, I.M.; Sofronis, P.; Nagao, A.; Martin, M.; Wang, S.; Gross, D.; Nygren, K. Hydrogen embrittlement understood. Metall. Mater. Trans. A 2015, 46, 2323–2341. [Google Scholar] [CrossRef] [Green Version]
- Venezuela, J.; Liu, Q.; Zhang, M.; Zhou, Q.; Atrens, A. A review of hydrogen embrittlement of martensitic advanced high-strength steels. Corros. Rev. 2016, 34, 153–186. [Google Scholar] [CrossRef]
- Depover, T.; Laureys, A.; Pérez Escobar, D.; Van den Eeckhout, E.; Wallaert, E.; Verbeken, K. Understanding the Interaction between a Steel Microstructure and Hydrogen. Materials 2018, 11, 698. [Google Scholar] [CrossRef] [Green Version]
- Herring, D.H. Hydroqen embrittlement. WFTI 2010, 13, 24–27. [Google Scholar]
- Birnbaum, H.K.; Sofronis, P. Hydrogen-enhanced localized plasticity—A mechanism for hydrogen-related fracture. Mater. Sci. Eng. A 1994, 176, 191–202. [Google Scholar] [CrossRef]
- Von Pezold, J.; Lymperakis, L.; Neugebeauer, J. Hydrogen-enhanced local plasticity at dilute bulk H concentrations: The role of H–H interactions and the formation of local hydrides. Acta Mater. 2011, 59, 2969–2980. [Google Scholar] [CrossRef]
- Wen, M.; Zhang, L.; An, B.; Fukuyama, S.; Yokogawa, K. Hydrogen-enhanced dislocation activity and vacancy formation during nanoindentation of nickel. Phys. Rev. B 2009, 80, 094113. [Google Scholar] [CrossRef]
- Zhong, L.; Wu, R.; Freeman, A.; Olson, G. Charge transfer mechanism of hydrogen-induced intergranular embrittlement of iron. Phys. Rev. B 2000, 62, 13938. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S. A conundrum for density functional theory. Science 2017, 355, 28–29. [Google Scholar] [CrossRef]
- Skylaris, C.-K. A benchmark for materials simulation. Science 2016, 351, 1394–1395. [Google Scholar] [CrossRef]
- Sedghamiz, E.; Khashei, F.; Moosavi, M. Linear tricationic ionic liquids: Insights into the structural features using DFT and molecular dynamics simulation. J. Mol. Liq. 2018, 271, 96–104. [Google Scholar] [CrossRef]
- Saha, S.K.; Ghosh, P.; Hens, A.; Murmu, N.C.; Banerjee, P. Density functional theory and molecular dynamics simulation study on corrosion inhibition performance of mild steel by mercapto-quinoline Schiff base corrosion inhibitor. Phys. E Low-Dimens. Syst. Nanostructures 2015, 66, 332–341. [Google Scholar] [CrossRef]
- Khnifira, M.; Boumya, W.; Attarki, J.; Mahsoune, A.; Sadiq, M.; Abdennouri, M.; Kaya, S.; Barka, N. A combined DFT, Monte Carlo, and MD simulations of adsorption study of heavy metals on the carbon graphite (111) surface. Chem. Phys. Impact 2022, 5, 100121. [Google Scholar] [CrossRef]
- Tateyama, Y.; Ohno, T. Stability and clusterization of hydrogen-vacancy complexes in α-Fe: An ab initio study. Phys. Rev. B 2003, 67, 174105. [Google Scholar] [CrossRef]
- Kholtobina, A.S.; Pippan, R.; Romaner, L.; Scheiber, D.; Ecker, W.; Razumovskiy, V.I. Hydrogen Trapping in bcc Iron. Materials 2020, 13, 2288. [Google Scholar] [CrossRef]
- Hayward, E.; Fu, C.-C. Interplay between hydrogen and vacancies in α-Fe. Phys. Rev. B 2013, 87, 174103. [Google Scholar] [CrossRef]
- Ma, Y.; Shi, Y.; Wang, H.; Mi, Z.; Liu, Z.; Gao, L.; Yan, Y.; Su, Y.; Qiao, L. A first-principles study on the hydrogen trap characteristics of coherent nano-precipitates in α-Fe. Int. J. Hydrog. Energy 2020, 45, 27941–27949. [Google Scholar] [CrossRef]
- Counts, W.; Wolverton, C.; Gibala, R. First-principles energetics of hydrogen traps in α-Fe: Point defects. Acta Mater. 2010, 58, 4730–4741. [Google Scholar] [CrossRef]
- Arrhenius, S. On the reaction rate of the inversion of non-refined sugar upon souring. Z. Phys. Chem. 1889, 4, 135501. [Google Scholar]
- De Morton, M. Measurement of nitrogen diffusion in chromium by anelastic methods. J. Appl. Phys. 1962, 33, 2768–2770. [Google Scholar] [CrossRef]
- Gomer, R. Diffusion of adsorbates on metal surfaces. Rep. Prog. Phys. 1990, 53, 917. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Chen, C.; Jiang, R.; Qiu, P.; Li, Y. Migration of ion vacancy in hydroxylated oxide film formed on Cr: A density functional theory investigation. J. Phys. Chem. C 2012, 116, 25478–25485. [Google Scholar] [CrossRef]
- Dwivedi, S.K.; Vishwakarma, M. Hydrogen embrittlement in different materials: A review. Int. J. Hydrog. Energy 2018, 43, 21603–21616. [Google Scholar] [CrossRef]
- Du, Y.A.; Ismer, L.; Rogal, J.; Hickel, T.; Neugebauer, J.; Drautz, R. First-principles study on the interaction of H interstitials with grain boundaries in α-and γ-Fe. Phys. Rev. B 2011, 84, 144121. [Google Scholar] [CrossRef]
- Eberhart, M. Computational metallurgy. Science 1994, 265, 332–333. [Google Scholar] [CrossRef]
- Chen, Y.-S.; Lu, H.; Liang, J.; Rosenthal, A.; Liu, H.; Sneddon, G.; McCarroll, I.; Zhao, Z.; Li, W.; Guo, A. Observation of hydrogen trapping at dislocations, grain boundaries, and precipitates. Science 2020, 367, 171–175. [Google Scholar] [CrossRef]
- Oudriss, A.; Creus, J.; Bouhattate, J.; Conforto, E.; Berziou, C.; Savall, C.; Feaugas, X. Grain size and grain-boundary effects on diffusion and trapping of hydrogen in pure nickel. Acta Mater. 2012, 60, 6814–6828. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.; Fullea, J.; Andrade, M.; De Andres, P. Ab initio molecular dynamics simulation of hydrogen diffusion in α-iron. Phys. Rev. B 2010, 81, 132102. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Li, Y.; Chen, C.; Yu, H. Diffusion coefficient of hydrogen interstitial atom in α-Fe, γ-Fe and ε-Fe crystals by first-principle calculations. Int. J. Hydrog. Energy 2017, 42, 27438–27445. [Google Scholar] [CrossRef]
- Zhou, X.; Mousseau, N.; Song, J. Is hydrogen diffusion along grain boundaries fast or slow? Atomistic origin and mechanistic modeling. Phys. Rev. Lett. 2019, 122, 215501. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, L.; Liu, S. Rapid Hydrogen Transportation Along Grain Boundary in Nickel. Acta Metall. Sin. 2014, 27, 615–620. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C (wt%) | Si (wt%) | Mn (wt%) | P (wt%) | S (wt%) | Ni (wt%) | Cr (wt%) | Mo (wt%) | V (wt%) | Fe | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1# | 0.12 | 0.2 | 1.02 | <0.01 | 0.001 | 1.12 | 0.42 | 0.47 | 0.03 | R |
| 2# | 0.14 | 0.19 | 1.38 | <0.01 | 0.002 | 0.30 | 0.22 | 0.24 | 0.03 | R |
| YS/MPa | UTS/MPa | KV2 (−40)/J | |
|---|---|---|---|
| 1# | 792.5 | 843 | 219.3 |
| 2# | 804.5 | 844 | 189 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, X.; Mi, Z.; Yang, L.; Su, H. Application of DFT Simulation to the Investigation of Hydrogen Embrittlement Mechanism and Design of High Strength Low Alloy Steel. Materials 2023, 16, 152. https://doi.org/10.3390/ma16010152
Fan X, Mi Z, Yang L, Su H. Application of DFT Simulation to the Investigation of Hydrogen Embrittlement Mechanism and Design of High Strength Low Alloy Steel. Materials. 2023; 16(1):152. https://doi.org/10.3390/ma16010152
Chicago/Turabian StyleFan, Xiuru, Zhishan Mi, Li Yang, and Hang Su. 2023. "Application of DFT Simulation to the Investigation of Hydrogen Embrittlement Mechanism and Design of High Strength Low Alloy Steel" Materials 16, no. 1: 152. https://doi.org/10.3390/ma16010152